Search Thermo Fisher Scientific
Proteomics Sample Preparation Information

Optimized sample prep workflows
Because the proteome is so complex, there is no one standard method for preparing protein samples for analysis by mass spectrometry (MS). Protocols differ depending on sample type, experimental goals, and analytical method used. Many factors are considered when designing sample preparation strategies, including source, type, physical properties, abundance, complexity and cellular location of the proteins. Workflows that incorporate optimized cellular lysis, subcellular fractionation, depletion of high-abundance proteins or enrichment of select proteins, and mass tagging tools can all contribute to the accurate identification and quantitation of protein samples. In all cases, the quality and reproducibility of sample extraction and preparation significantly impact your research results.
Featured webinar
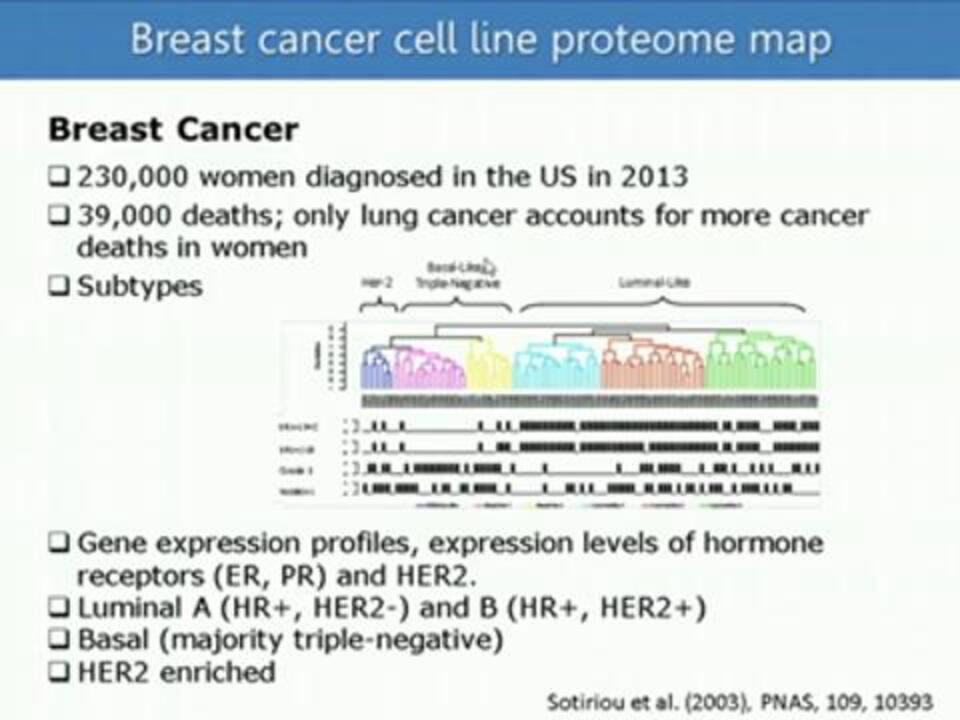
High-Coverage High-Throughput Characterization of Breast Cancer Cell Line Proteomes Using 10-Plexed TMT on an Orbitrap Fusion
Dr. Wilhelm Haas of Harvard University/Massachusetts General Hospital discusses the scientific and technical challenges that have prevented the "cancer proteome" from being a greater source of insight into cancer. He presents a workflow that combines the use of isobaric mass tags and advanced mass spectrometric techniques to quantitatively analyze almost 8000 proteins in 32 breast cancer cell lines in only 4.5 hours per cell line.
Mass spectrometry (MS) has become the method of choice for protein detection, identification, characterization, and quantitation. The accuracy, sensitivity and flexibility of MS instruments have enabled new applications in biological research, biopharmaceutical characterization and diagnostic detection. Proper sample preparation for MS-based analysis is a critical step in the proteomics workflow, because it can be both variable and time consuming. The quality and reproducibility of sample extraction and preparation significantly impact MS results.
Cell lysis is frequently the first step in protein extraction, fractionation and purification. Lysis can be accomplished either physically or by using reagents, though reagent-based approached are generally favored as they are faster and more reproducible. By using different buffers, detergents, salts and reducing agents, cell lysis can be optimized to provide the best possible results for a particular cell type or protein fraction. Lysis is often followed by stabilization to protect extracted proteins from degradation or artifactual modification. Depletion and enrichment strategies are often employed to remove high-abundance proteins of no analytical interest and isolate target proteins in the sample. Thermo Fisher Scientific offers a complete range of reagents, inhibitors, and depletion and enrichment kits to optimize your analyses.
Many protein research objectives are more easily achieved through the analysis of peptides rather than intact proteins. For these applications, proteins must be subject to a process of denaturation, reduction, alkylation, and digestion. Digestion can take place in solution or in gel depending on sample characteristics and whether gel electrophoresis is part of your normal workflow. Thermo Scientific denaturants, reducing agents, stains, and endoproteinases are of the highest grade to help you get the best possible results from your analyses.
Enrichment of specific target peptides and sample clean-up are required for successful analysis of low-abundance proteins or identification of post-translationally modified peptides. Enrichment for specific PTMs (e.g., phosphorylation, ubiquitination and glycosylation) is performed by affinity purification using PTM-specific antibodies or ligands. After peptide enrichment, salts and buffers can be removed using either graphite or C-18 tips or columns, and detergents can be removed using affinity columns or detergent-precipitating reagents. Dilute samples can also be concentrated using concentrators of varying molecular weight cutoff (MWCO) ranges. Thermo Fisher Scientific offers complete kits for sample enrichment and a variety of tips, columns, and resins for sample cleanup.
Quantitative analysis is used throughout proteomics research. During the discovery phase, relative quantitation is frequently use to locate proteins that show statistically significant changes in abundance between sample classes or over time and thus are more likely to be of scientific interest. Quantitative analyses are later used to verify and validate the putative biomarkers identified during discovery. Label-free quantitation based on comparison of ion peak intensities or spectral counts is the simplest method of quantitation, but it is more susceptible to errors due to sample and experimental variations, and experimental bias. Other techniques can be employed to enhance the speed, accuracy, or power of quantitative protein analyses, including:
- Metabolic (in vivo) labeling with stable isotopes
- Isotopic tags
- Isobaric mass tags
- Spiked heavy peptides
Thermo Fisher Scientific offers a complete range of products to enhance your quantitative proteomics analyses including SILAC (stable isotope labeling by amino acids in cell culture) kits, TMT tandem mass tags, and stock or custom heavy peptide standards.

Clinical & Translational Resource Library
Access a targeted collection of scientific application notes, case studies, videos, webinars, and white papers for clinical and translational research.
Support