Search Thermo Fisher Scientific
- Contact Us
- Quick Order
-
Don't have an account ? Create Account
Search Thermo Fisher Scientific
With proteins, form is function. To understand protein function and mechanism of action, it is essential to determine protein complex assembly and structure. We lead the way in accelerating protein structure-function studies with Integrative Structural Biology solutions, which bring together complementary techniques such as mass spectrometry and cryo-electron microscopy (cryo-EM). Together they can solve the structure of large dynamic complexes.
An MS approach to explore protein folding, weak protein–protein interactions, and other rapid processes at a residue-level resolution on the microsecond timescale.
Download the new Integrative Structural Biology flyer titled "Higher precision 3D analysis from structure to function" that highlights how various mass spectrometry (MS) techniques can be combined with cryo-EM to provide unique insights beyond protein structures. The flyer provides an overview of the various opportunities for combining MS with cryo-EM to assist researchers to go beyond structure alone and understand how protein functions. It further compares the strengths of all of the techniques discussed in a combined cryo-EM and MS workflow, whether for screening, solving structures or understanding protein function.
Technology developments in MS have given rise to several applications of structural biology, both at the single protein and protein complex level. The primary advantages of MS-based techniques are the ability to perform experiments at proteome scale, to analyze proteins in their native biological state, and to reduce the minimum required sample size.
Mass spectrometry (MS) is commonly used to determine both the primary and higher-order structures of proteins. New advances in MS technologies, combined with chemical modification and proteolysis strategies, allow the study of both single proteins and protein complexes as well as further exploration of protein structure and even structural dynamics.
Thermo Scientific Orbitrap MS solutions enable highly specific and sensitive workflows that allow you to analyze samples with increasing analytical depth, delivering information to accelerate the path from structure to function.
Peptide sequencing via mass spectrometry, when performed using a bottom-up approach, is a useful and easy tool for obtaining information about primary protein structure. Such information helps elucidate the identity of that protein, or even the identities of several proteins involved in larger protein complexes.
Hydrogen deuterium exchange mass spectrometry (HDX-MS) measures the conformational changes of proteins or protein-protein interactions in protein complexes. Amide protons found within close inter- or intra-molecular contact areas often form hydrogen bonds and have different exchange rates relative to more accessible regions of the complex. By monitoring such exchanges, a protein's noncovalent structure (alone or in complex) is better understood.
Protein structure data can be obtained independently or simultaneously as part of protein interaction studies using crosslinking mass spectrometry (XL-MS). In such analysis, XL-MS is often combined with high-resolution techniques (e.g., cryo-electron microscopy (cryo-EM), X-ray crystallography). Such technologies help determine protein region distance constraints via 3-D structure information or topology. XL-MS can also be used to identify protein complexes.
A limited proteolysis approach on single proteins, coupled to MS, can yield answers about the higher-order structure of proteins and their folded states. This approach can also be used to probe the quaternary structure of protein complexes. The formation of an interface between a protein and another macromolecule will protect otherwise accessible sites from protease cleavage, providing information about the residues that form that interface.
Protein interactions with small molecules, which are termed protein-ligand interactions, are involved in numerous biological functions, from protein transcription to translation and signal transduction. Knowing which conformational changes are induced upon small-molecule binding, and which molecules can bind target proteins better than their own natural ligands, are key goals of biotherapeutic design.
Information on protein-ligand complex interactions, such as stoichiometry and dissociation constant, is often obtained using MS. Protein-ligand complexes are analyzed in the mass spectrometer in their native biological states. Mass measurement of the complexes can confirm the presence of protein-ligand interactions, while stoichiometry is determined by taking mass measurement with and without protein-ligand interactions.
Hydrogen deuterium exchange mass spectrometry (HDX-MS) aids in localization of protein-ligand interaction sites. Protein-ligand complexes are dissolved in a solution of D2O, which enables the labile amide protons of the protein to exchange with deuterium. Typically, areas of protein-ligand interaction exchange at a much slower rate than open regions exposed to D2O. MS is then used to determine proton exchange rates, helping to localize interaction sites and elucidate their structural information.
Crosslinking mass spectrometry (XL-MS) is used to determine protein-ligand binding sites. This process is initiated by using photoaffinity labels to photo-crosslink proteins with ligands. Samples are then enriched, digested, and analyzed by MS for ligand modifications on the protein.
Many cellular processes are regulated by precise amounts of specific protein complexes, ligands, or enzymes. Protein stoichiometry aims to measure the exact amounts of the individual components of these protein complexes, which is a requirement for fully understanding their overall function.
Quantitative peptide‐based MS approaches can be used to determine protein stoichiometries in protein complexes, the first step in elucidating protein complex structures. Alternatively, targeted approaches with known amounts of isotopically labeled standards of the protein/peptide in question can be spiked in to gain an accurate measurement of these protein complexes.
The direct introduction of protein complexes in their native biological state enables analysis of protein-protein and protein-ligand complexes. Mass measurements coupled to protein identifications confirm the precise stoichiometry of these complexes. This information is the crucial first step in protein complex structure determination.
Crosslinking mass spectrometry (XL-MS) can be used to determine the binding stoichiometry of individual protein complexes. This is advantageous when dealing with labile proteins and protein complexes. Typically, zero length or non-specific crosslinkers are used to preserve the interacting partners for measurement of protein stoichiometry.
A protein's biological activities are dictated by its interactions with other proteins. Protein-protein interactions control cellular processes including protein modification, transport, folding, signaling, and cell cycling. In order to fully understand protein function, proteins should be studied in the context of their interactions with other proteins.
Native mass spectrometry (MS) plays a crucial role in generating information on protein-protein interactions, confirming protein identity, and elucidating heterogeneity data. Solvent manipulation during protein complex introduction into the mass spectrometer can reveal additional information. For example, varying solvent pH or adding organic additives can separate protein subcomplexes, which are then fragmented to confirm identity. Native MS can also give stoichiometry information on the proteins involved in protein-protein interactions.
In crosslinking mass spectrometry (XL-MS), chemical crosslinkers are used to join components of interacting complexes in order to maintain their original interaction. A bottom-up proteomics MS approach is then used. One advantage of XL-MS is that only a small sample size is required, with analysis performed directly at the proteome level. XL-MS also enables protein interactions to be examined at the physiological state of an organism, generating information that is biologically relevant.
Hydrogen deuterium exchange mass spectrometry (HDX-MS) helps localize protein-protein interaction sites. Typically, areas of protein-protein interaction have hindered exchange rates between amide protons and D2O compared to regions with direct exposure to D2O. Because of this difference, protein-protein interaction sites can be localized using proton exchange. MS then identifies those sites. HDX experiments are usually performed on proteins using conditions that are native to their biological state.
Regardless of the question, the mainstay of proteomics is protein identification. In current laboratory practice, protein identification and mass spectrometry (MS) are nearly synonymous because MS allows for protein analysis from any sample of varying complexity, is high-throughput, and is quantitative. With MS, proteins can be identified at the intact (top-down) protein level or by using the more popular strategy, bottom-up proteomics. With the latter strategy, proteins are enzymatically digested down to their peptide components, then analyzed at the peptide level.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is the primary workflow for most researchers when performing protein identification. Proteins are enzymatically digested to their peptide components, then analyzed by LC-MS/MS. The resulting sequence data are used to determine the original protein components of the sample. For single proteins, sequence usually confirms identity. When protein complexes are involved, this top-down proteomics approach determines the individual proteins that are part of the complex.
Protein identification can be achieved without prior digestion at the intact protein level using gas phase fragmentation. This approach enables combinations of posttranslational modifications (PTMs) to be localized, permitting higher sequence coverage for individual proteins and characterization of proteoforms. Additional information, such as degradation products and sequence variants, can also be obtained within this type of experiment.
The majority of proteins undergo some level of posttranslational modification (PTM) on their amino acid residues, which influences their biological function in processes such as catalysis, cell-cell signaling, and degradation. Mapping PTM sites on individual protein subunits provides information on subunit function and regulation. Within the protein complex, mapped PTMs can also help predict their partner protein interactions.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is the most frequently used approach to identify and localize PTMs on proteins and protein complexes. Multiple fragmentation methods are typically implemented during bottom-up PTM workflows to ensure complete protein characterization.
Middle-down proteomics (low-resolution studies of protein structure) involves limited proteolysis of proteins. This method permits sequence coverage of larger peptides in their native configuration, which is useful when characterizing protein-protein interactions or when probing for labile regions during protein reconstruction work for crystallization studies.
The primary advantage of top-down MS analysis is its ability to identify and localize combinations of PTMs. Using this approach, proteins are analyzed without prior digestion of their corresponding peptide species. Top-down MS analysis can involve single or multiple proteins from complex mixtures. Additional information, such as degradation products and sequence variants, is often obtained during these experiments.
Native MS is used to ascertain the expected patterns of and degree of PTM on a protein. It can also provide information on the relative abundance of modifications (e.g., glycoforms) that are present at a particular site. Due to the inherent heterogeneity and variation of attached PTMs, native MS is often performed using high-resolution, accurate-mass spectrometry.
HDX-MS can be used to study protein conformational changes induced by chemical modifications. In HDX experiments, deuterium exchange rates of unmodified and modified proteins are compared. By monitoring deuterium uptake, information is obtained on how the modification has affected the protein under study.
Specific ligands can be chemically bound to a solid support, allowing for the enrichment of certain classes of biological molecules as a function of their chemical affinity. Example molecules include phosphate, glycosyl, and ubiquitin groups. Through the use of binding ligands, specific PTMs and peptides are affinity purified for eventual MS analysis.
Obtain maximum insights on your most complex molecules and biological systems, from whole proteome profiling and quantitation, structural characterization to multiplexed single-cell proteomics. With new innovations that deliver the ultimate flexibility in experimental scope, the Thermo Scientific Orbitrap Ascend Structural Biology Tribrid Mass Spectrometer accelerates your path to new, impactful results, so you can drive your science beyond today’s discovery.
Obtain maximum quantitative insights from untargeted proteome profiles to targeted proteomics experiments with industry leading single-cell sensitivity and extraordinary accuracy, precision and simplicity. With curated workflows that deliver greater usability, the Thermo Scientific Orbitrap Exploris 480 Mass Spectrometer accelerates your path to large-scale studies, delivering proven high data quality and time savings, so you can go beyond faster to actionable outcomes.
Solving the structure of large dynamic complexes requires integrating several complementary techniques, such as mass spectrometry and cryo-EM density maps, in an approach that is known as integrative structural biology.
Advances in mass spectrometry combined with recent developments in cryo-EM sample preparation, data collection and image processing make it possible for a reliable and complete structure to be solved for macromolecular complexes composed of components like proteins, post-translational protein modifications, DNA, RNA and lipids. Cryo-EM, particularly single partical analysis (SPA) is becoming an essential technique for structural determination of protein complexes.
Biomolecular mass spectrometry has significantly advanced and impacted the field of structural biology. At the intact protein level, native MS enables the study of protein assemblies in their native state through the analysis of non-covalent protein-protein and protein-ligand complexes. At the peptide level, LC-MS/MS analysis of protein proteolytic digests helps determine the amino acid sequence of proteins, allowing their subunits to be identified from a proteome database.
Thermo Scientific Orbitrap MS solutions enable both peptide and protein-centric strategies that deliver insights into a multitude of biochemical and structural properties. Armed with superior quality data from proven HRAM Orbitrap systems, you can confidently analyze samples with increasing analytical depth, and deliver information that accelerates the journey from structure to function.
In collaboration with the editors of Science Magazine, we invite you to download this comprehensive booklet, containing articles from the Science family of journals, perspectives on integrative structural biology, and interviews with leading researchers.
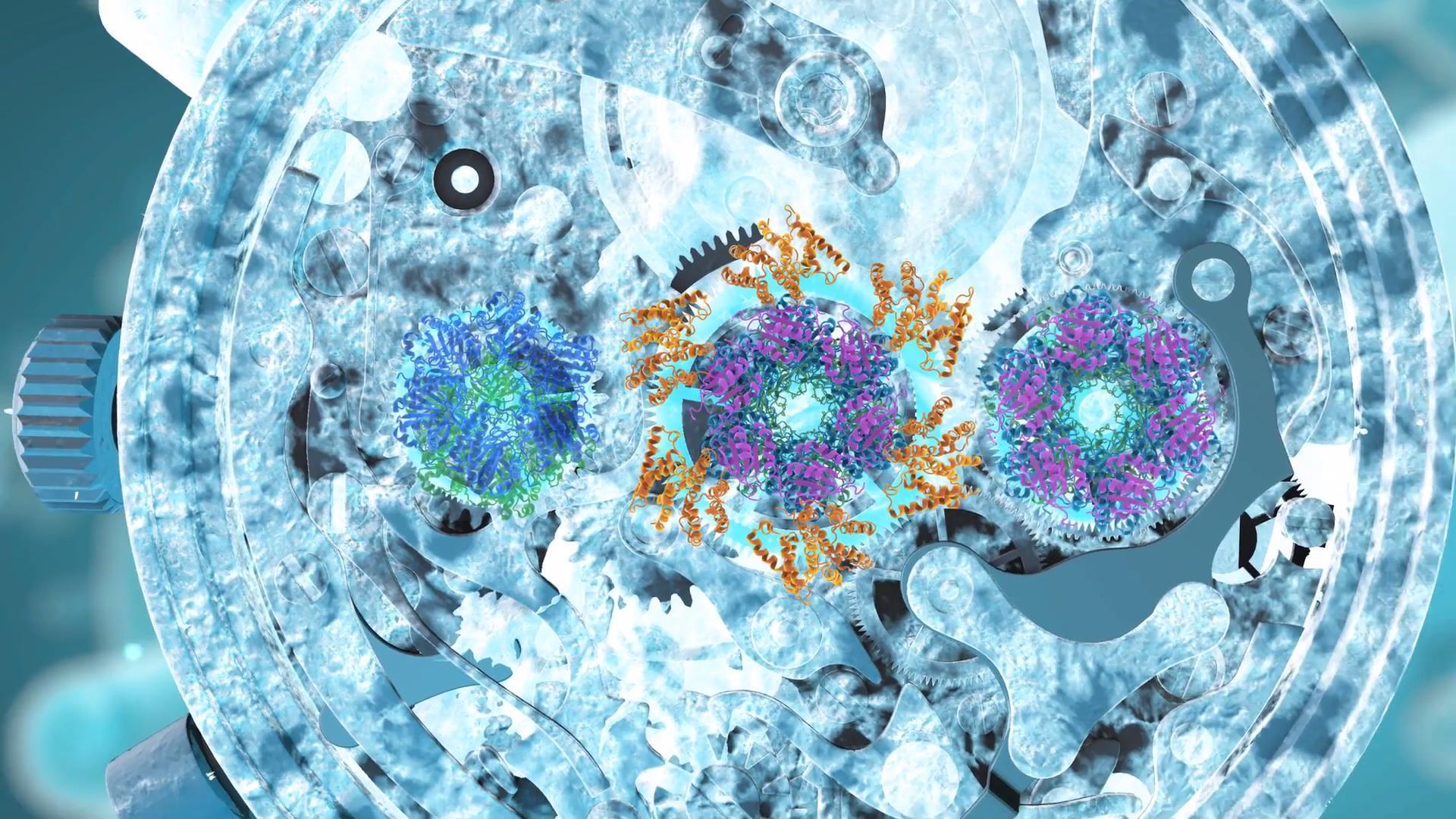
We describe the preparation of stoichiometrically well-defined assemblies of KaiCB and KaiCBA, as monitored by native mass spectrometry, allowing for a structural characterization by single-particle cryo-electron microscopy and mass spectrometry.
Watch the on-demand recordings to learn how combining mass spectrometry and cryo-EM allows structural biologists to achieve unparalleled results in their quest to understand macromolecular functions.













")









")






