Search Thermo Fisher Scientific
PCR方法—常用的10种策略

表 1.常用PCR方法及其核心优势。
产品视频

热启动PCR常用于增强PCR扩增的特异性。该方法主要利用抗体、亲合配体、适体或化学修饰物等酶修饰酶,来抑制室温下的DNA聚合酶的活性。这种修饰使得在PCR体系配制阶段引物与模板、引物与引物之前的结合能力降低,从而避免了非特异性扩增。由于DNA聚合酶在室温下的活性被抑制,所以,热启动技术为在室温下配制多个PCR反应体系提供了极大的便利,,且不会影响特异性和扩增能力(了解关于 DNA 聚合酶特点的更多信息)。
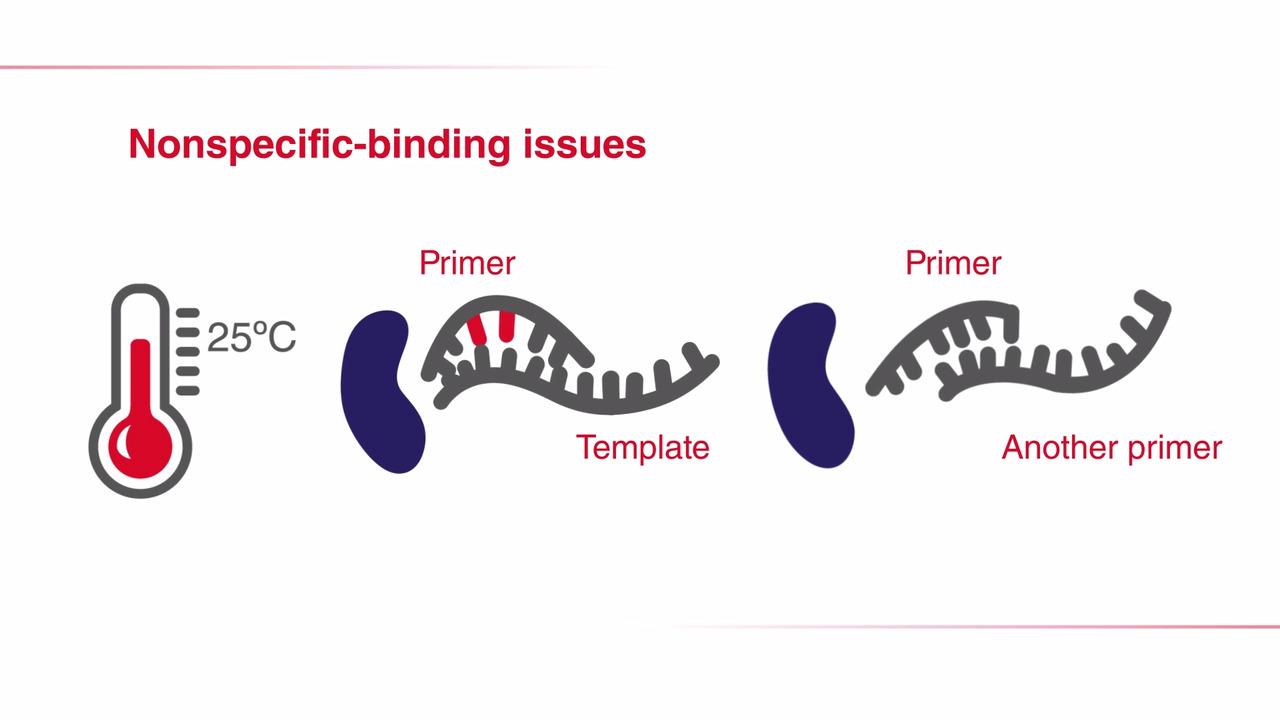
当反应体系配制好后,在反应初始加热阶段或“热启动”阶段,酶修饰物在高温下(通常高于90 ℃)被释放,使得DNA聚合酶被激活(图1)。具体激活时间和温度取决于DNA聚合酶以及热启动修饰物的性质。对某些DNA聚合酶而言,有时激活和起始变性步骤可以合并为一步。

图 1.基于抗体热启动技术的DNA聚合酶。
另一种提高PCR反应特异性的方法是调整PCR循环的参数。在降落PCR中,前面几个循环的退火温度设定为比引物的最高熔解温度(Tm)再高几度[1,2]。较高的温度有助于避免产生引物二聚体及非特异性引物-模板复合物的形成,因此可减少不希望出现的扩增。因此,在PCR起始阶段提高退火温度,可减少非特异性PCR产物,增加特异性扩增(了解关于 PCR 退火步骤的更多信息)。
需要注意的是,虽然较高的退火温度能够防止引物二聚体形成和非特异性引物结合,但同时也可能加剧引物与目的序列的解离,从而降低PCR得率。为克服这一问题,在最初几个循环中,通常会将每个循环的退火温度降低1°C,以获得足量的目标扩增子。一旦退火温度达到或“降落”至最佳温度(通常比最低引物Tm低3-5°C),剩下的循环都维持此退火温度。通过这种方法,在PCR过程中,所期望的PCR产物得到有选择性的增加,同时保证很少或不发生非特异性扩增(图 2)。

图 2:降落PCR该方法通过采用高于最佳退火温度的起始温度,之后随着循环逐渐降温(黑线),直至达到最佳退火温度,来提高特异性(黄色曲线)。退火温度不断优化,目标扩增子的得率(绿色曲线)不断累积。
巢式PCR是标准PCR的一种演变,其增强了反应特异性和目标扩增子的产量[3]。在此方法中,需要设计两对PCR引物:一对(外引物)在目标扩增区域的侧翼,另一对(巢式引物)对应于待扩增的DNA区域。其中,外引物用于第一轮PCR,以扩增含有延伸侧翼区域的区域。随后,巢式引物用于第二轮PCR,并以第一轮PCR产物为模板(图 3)。

图 3.巢式PCR
如果第一对引物(外引物)的错配导致非特异性产物被扩增,相同的非特异性区域被第二对引物识别并继续扩增的可能性非常小,所以通过第二对引物的扩增,PCR的特异性得到了提升。进行两轮PCR的一个优势在于:有助于从有限的起始DNA中扩增得到足量的产物。
在快速PCR中,通过缩减PCR步骤所需时间来完成更快的扩增,且不会影响扩增产量和效率。快速循环条件尤其适用于具有高扩增能力的DNA聚合酶,这类聚合酶在每个结合中可引入更多的核苷酸(了解关于 合成能力的更多信息)。高合成能力 Taq 聚合酶所需的延伸时间仅占低合成能力Taq聚合酶所需时间的1/2至1/3,却能维持较高的扩增效率(图4)。此外,如果引物的退火和延伸温度相差无几,则可将它们合并为一步,以进一步缩短PCR时间。这一过程也被称为 两步PCR法。

图 4.使用低合成能力和高合成能力DNA聚合酶扩增来自人类gDNA 3.8 kb片段的结果对比。使用两步PCR实验方案(合并退火和延伸步骤)。
当使用低合成能力的 Taq 聚合酶时,如Taq聚合酶,快速循环条件可能适用于 <500 bp左右的短片段。扩增如此大小的片段通常不需要延长聚合时间,因此可缩短PCR方案中的延伸步骤时间。为确定最短延伸时间,同时又不损失产物得率,可采用一系列延伸时间递减的方式(几秒)优化PCR。每个目的片段和引物对都可能会产生变化结果,所以需要在特定条件下对快速PCR进行优化。
快速PCR的另一种调整方式是缩短变性时间,将变性温度提高至98°C。使用这种策略时应注意,非高度热稳定的酶在这种高温环境下易于变性(了解关于 DNA 聚合酶热稳定性的更多信息)。
表 2.使用低合成能力DNA聚合酶实现快速PCR时所用的反应参数。
| 参数 | 快速PCR的优化 |
|---|---|
| 扩增子长度 | <500 bp |
| 变性时间 | 降低 |
| 变性温度 | 升高 |
| 延伸时间 | 降低 |
直接PCR是指直接从样品扩增目标DNA,无需进行核酸分离纯化。直接PCR中,在高温变性阶段,诸如细胞、组织等材料在特殊的缓冲液中被裂解,释放出DNA。因此这种方法简化了实验流程,减少了动手操作时间,同时可避免纯化步骤DNA的损失(图 5)。

图 5.常规PCR和直接PCR对比。
推荐使用具有 高合成能力 的DNA聚合酶用于直接PCR扩增。细胞碎片、蛋白、脂质和多糖也随DNA一起被释放到裂解液中,它们会抑制PCR反应。而具有高合成能力的DNA聚合酶能够耐受这类抑制剂,使直接PCR扩增成为可能。具有高合成能力的酶通常具有更高的灵敏度,因此可从未纯化的样品中成功扩增微量DNA。
具有高GC含量(>65%)的DNA模板由于G和C碱基间的强氢键影响,比较难以扩增。富含GC的序列同时也涉及二级结构。因此,富含GC的序列可导致DNA聚合酶沿模板扩增时“卡顿”并干扰DNA合成。
为了扩增高GC含量的片段,双链模板必须解离,以便引物与模板结合,并使DNA聚合酶能够读取到序列。为了克服强GC相互作用,最常用的方法是使用DMSO等 PCR添加剂或辅助溶剂 来帮助DNA变性(图 6A)。然而,这些试剂通常会降低引物的 Tm,所以退火温度也需进行相应的调整。
高合成能力的DNA聚合酶由于与模板的结合能力更强,有利于完成高GC含量PCR(图 6B)。超高热稳定性DNA聚合酶也有利于高GC含量PCR,因为较高的变性温度(如,使用98°C代替95°C)可能会促进双链解离和PCR扩增(了解关于 PCR循环的更多信息)。

图 6.不同GC含量人类gDNA区域的扩增。(A)使用低合成能力DNA聚合酶扩增GC含量为76%的~0.8 kb靶标。增加DMSO添加剂的用量,可提高特异性。(B) 使用 高合成能力DNA聚合酶扩增GC含量不同的七个片段。只有GC含量为70%和76%的片段使用GC增强剂。
多重PCR可在同一PCR反应管中同时扩增多个不同的片段。多种PCR不仅意味着节省时间、试剂和样品,还能够同时对比多个扩增子(图 7)。

图 7.单个PCR和多重PCR的比较。在单个PCR中,每个反应使用一个引物对扩增一个目的片段。而在多重PCR中,每个反应使用多个引物对扩增多个目的片段。
当一个PCR管中有多个引物对时,如在多重PCR中,因无法仅针对一个引物对或目的片段进行反应优化,而是要考虑到所有引物和靶标,所以可能会出现非特异性扩增和效率降低。因此,为尽量减少由非特异性扩增导致的错配,应对引物进行精心设计。首先,引物序列应尽可能与其目的序列一一对应,并且所有引物的 Tm相差不应超过5°C。在多重PCR开始前,应利用单个PCR反应验证每个引物对的特异性和扩增效率。此外,扩增子应具有不同的大小,从而能够通过凝胶电泳对其进行分离鉴定。除了引物设计和扩增子大小,使用热启动DNA聚合酶和专为多重PCR设计的缓冲液也将有助于获得成功的PCR结果和提高反应特异性(图8)。

图 8.通过凝胶电泳对比单个和多重PCR结果。该实验中使用Invitrogen™ Platinum™ 多重PCR预混液。
长片段PCR通常是指扩增大于5kb的DNA片段。长片段PCR传统上使用 Taq DNA 聚合酶(用于快速延伸)和高保真酶(用于提高准确性)的混合物。
随着具有 高合成能力的高保真DNA聚合酶被发明出来,现在能够在更短的时间内实现更准确的长片段PCR。通过在DNA聚合酶中设计一个较强的DNA结合结构域,从而使其能够在短时间内扩增长片段(如,来自gDNA的 >20 kb 片段),实现高合成能力(图9, 表 3)。此外,极高的保真度(如, Taq 聚合酶保真度的 >100倍)还有助于确保长片段扩增的低错误率(表 3)。(了解关于 DNA聚合酶特点的更多信息。)

图 9.使用 高合成能力DNA聚合酶扩增长片段。从人类gDNA样品获得15 kb和30 kb片段的特异性扩增。
表 3.在长片段PCR和克隆中使用 高合成能力高保真DNA聚合酶 的优势。
DNA聚合酶的高合成能力可显著缩短长片段PCR的反应时间(在本例中,时间缩短了一半),而高保真度可减少筛选含正确插入片段克隆的工作量。
| Taq 和校正DNA 聚合酶的混合物。 | 高合成能力高保真DNA聚合酶 | |
|---|---|---|
| 延伸速度 | 60秒/kb | 30秒/kb |
| PCR时间 (20 kb目的片段,30个循环) | 约10.5小时 | 约5.2小时 |
| 保真度 (相对于Taq DNA聚合酶) | 5x | >100倍 |
| 克隆错误率 (20kb,30-循环PCR) | 平均每个克隆的错误率为2.5% | 每4个克隆中,有1个克隆含有一个错误 |
当扩增>10 kb的目的片段时,应根据以下5个关键点对PCR方案进行优化:
- 确保使用高质量、高纯度的DNA样本。
- 如果DNA聚合酶的热稳定性较低,则需要使用更多量的酶,以弥补因延长循环时间导致的活性损失。
- 降低退火和延伸步骤的温度,有助于引物结合。
- 适当延长PCR步骤的持续时间,有助于模板DNA的完全解离及引物的结合。
- 适当延长PCR延伸时间,可确保目标区域的全长复制。
反向PCR起初设计用于确定邻近未知区域的序列。它有助于研究基因的启动子序列;致癌性染色体重排,如基因融合、易位和转座;以及病毒基因整合。该方法之所以被称为反向PCR,是因为引物设计用于向两边延伸而不像常规PCR中朝着彼此延伸。[4,5]。如今,反向PCR常被用于 定点突变 ,复制一个具有预期突变的质粒。
在研究基因组DNA未知序列的传统工作流程中,首先进行限制性酶切消化和连接,再进行反向PCR,随后对PCR扩增子进行测序。对于gDNA消化,需选用一种限制性内切酶进行酶切,以获得长度合适且能够自我连接的片段。同时,选定的限制性内切酶不可剪切已知序列,从而使连接发生于侧翼未知序列之间。使用低浓度的酶切DNA片段优化连接步骤,使其倾向于自我连接而非多片段连接(即形成连环体)。
完成自我连接后,从DNA的已知区域启动反向PCR。所获得的扩增子每个末端都含有部分已知DNA序列。随后,可从末端开始对这些扩增子进行测序,检测上述已知序列的相邻区域(图 10)。

图 10.用于扩增和鉴定邻近未知序列的反向PCR。
序列的扩增程度(得率)取决于模板起始量,PCR常用于对样品中的DNA进行定量,其中,最常见的应用是 基因表达定量。终点PCR方法虽然可行,但它存在一重大缺点,即需要通过凝胶电泳确定得率,从而限制了检测灵敏度。此外,定量是在PCR末期进行的,而此时的扩增已达到平台期(图11),因此,DNA凝胶染色强度无法与DNA起始量呈线性相关。尽管如此,若在到达平台期之前通过终点PCR对基因表达进行半定量分析,可使用连续稀释的DNA样品作为起始物,或收集指定PCR循环的扩增子,并根据凝胶染色强度估计基因表达量[6,7]。

图 11.PCR的扩增曲线或反应动力学。在终点PCR中,在扩增到达平台期时对扩增子进行检测。而在实时荧光定量PCR中,是在指数增长期对扩增子进行定量。
直到1993年,Higuchi等报道称使用荧光信号对PCR扩增进行实时监测,才克服了终点PCR定量的局限性[8]。这一技术为我们今天所熟知的 定量PCR(qPCR)奠定了基础。1997年,第一款qPCR仪进入市场[9,10],使PCR能够准确定量基因表达和拷贝数。qPCR依靠对指数期目的片段扩增荧光信号的实时监测(图 11),克服了终点PCR定量的缺点。尽管qPCR能够定量检测相对和绝对基因表达,但是其检测能力限制了定量性能。
20世纪90年代与实时荧光定量PCR同时开发的 数字PCR (也称为极限稀释PCR)实现了真正的DNA样品绝对定量[11-13]。在数字PCR中,是将高度稀释的DNA样品分配到多区室芯片中,使每个区室最多含有一个拷贝的靶标。然后,对每个区室内的扩增进行检测,获得阳性或阴性结果(分别为1或0个模板拷贝;即, "数字" 结果)。最后,使用统计模型(泊松分布),根据阴性反应部分确定样品的拷贝数,无需定量已知样品(标准品)(图12)。除了基因表达和拷贝数定量,数字PCR还适用于区分低频等位基因、病毒滴定以及下一代测序文库的绝对定量等应用。

图 12.利用数字PCR进行绝对定量的一般工作流程。
总之,改进的PCR实验方案和改进的DNA聚合酶旨在改善PCR扩增的结果。虽然PCR的基础概念并未发生改变,但新型PCR方法将继续推动和简化分子生物学研究。
参考文献
- Don RH, Cox PT, Wainwright BJ (1991) 'Touchdown' PCR to circumvent spurious priming during gene amplification.Nucleic Acids Res19(14):4008.
- Hecker KH, Roux KH (1996) High and low annealing temperatures increase both specificity and yield in touchdown and stepdown PCR.Biotechniques20(3):478–485.
- Haff LA (1994) Improved quantitative PCR using nested primers.PCR Methods Appl3(6):332–337.
- Ochman H, Gerber AS, Hartl DL (1988) Genetic applications of an inverse polymerase chain reaction.Genetics120(3):621–623.
- Pavlopoulos A (2011) Identification of DNA sequences that flank a known region by inverse PCR.Methods Mol Biol772:267–275.
- Raeymaekers L (1999) General Principles of Quantitative PCR.In: Kochanowski B, Reischl U (editors), Quantitative PCR Protocols.Totowa, NJ: Humana Press. pp. 31–41.
- Siebert PD (1999) Quantitative RT-PCR.In: Kochanowski B, Reischl U (editors), Quantitative PCR Protocols.Totowa, NJ: Humana Press. pp. 61-85.
- Higuchi R, Fockler C, Dollinger G (1993) Kinetic PCR analysis: real-time monitoring of DNA amplification reactions.Biotechnology (N Y)11(9):1026–1030.
- Wittwer CT, Ririe KM, Andrew RV (1997) The LightCycler: a microvolume multisample fluorimeter with rapid temperature control.Biotechniques22(1):176–181.
- Applied Biosystems, Inc. (1997) Relative quantitation of gene expression: ABI PRISM 7700 Sequence detection system.User Bulletin #2: Rev B part 4304859B, 1–36.
- Sykes PJ, Neoh SH, Brisco MJ et al.(1992) Quantitation of targets for PCR by use of limiting dilution.Biotechniques13(3):444-449.
- Kalinina O, Lebedeva I, Brown J et al.(1997) Nanoliter scale PCR with TaqMan detection.Nucleic Acids Res25(10):1999–2004.
- Vogelstein B, Kinzler KW (1999) Digital PCR.Proc Natl Acad Sci U S A 96(16):9236–9241.
仅供科研使用,不可用于诊断目的。