Search Thermo Fisher Scientific

DNAポリメラーゼは、新たなDNA鎖合成において主要な役割を担っているため、PCRにとって必要不可欠な構成要素です。したがって、PCR法を広範なアプリケーションに適用するためには、この酵素の特性について理解したうえ、その改良を進めることが重要です。PCRプロトコル開発の初段階でTaq DNAポリメラーゼを使用したことで、特異性、耐熱性、フィデリティ、およびPCR酵素の処理能力が著しく向上しました。DNAポリメラーゼのこうした特性は、以下のセクションで述べるようにPCRを改善する方向に進化してきました。

非特異的増幅は、PCRにおける収量および感度に劇的な影響を及ぼす可能性があり結果の解釈およびダウンストリームアプリケーションの成果が損なわれる可能性があります。よって、非特異的増幅はPCRの大きな障害の1つと考えられています。DNAポリメラーゼが、プライマーがミスプライムした標的やプライマーダイマーを伸長してしまうことは度々起こります。このようなことが、非特異的増幅の一般的な原因です。非特異的増幅を減らす方法の1つは、反応液を氷上でセットアップすることです。これによりDNAポリメラーゼ活性を低く保つことができますが、それでもなお、PCR開始前に望ましくない産物が合成されてしまう可能性はあります。もう1つの解決策は、DNAポリメラーゼを添加するタイミングを、初回サイクルのアニーリングステップまで遅らせることです。この手法は、最初の変性ステップ(90°C以上)を経て初めて増幅をスタートできるため、「ホットスタート」と呼ばれています。
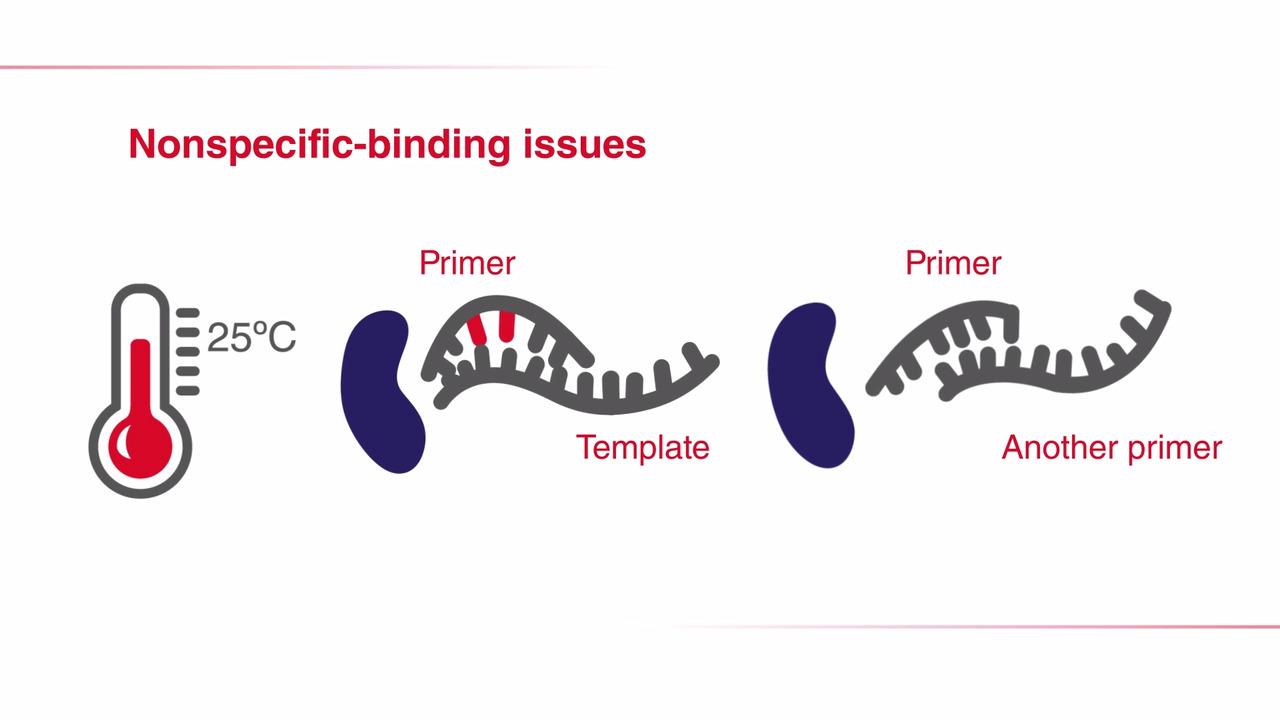
手作業でのホットスタート手順は、特異性の改善に効果的ではあるものの、手間が掛かるばかりか、サンプルの汚染および再現性低下のリスクが増大します。1994年に、真のホットスタート特性を持つTaq DNAポリメラーゼが開発されました[1、2]。このポリメラーゼには特異的な抗体が結合しており、反応を室温でセットアップする間、その活性が阻害されます。最初の高温変性ステップ(例えば、90°Cを超える)で、この結合した抗体が外れ、DNAポリメラーゼが活性化されます(図1)。

図1. 抗体を用いたホットスタートDNAポリメラーゼ。反応中に活性化され、PCRの特異性を高める。
変性ステップは、反応液セットアップ中に起こることがあるプライマーのミスアニールおよびプライマーダイマーを分離させます。その結果、後続するアニーリングおよび伸長ステップにおいて、望ましくない産物がDNAポリメラーゼによって増幅することを防ぐことができます。ホットスタートDNAポリメラーゼは、このように、非特異的増幅を低減し、収量を増大させ、簡便な室温でのセットアップが可能となるためハイスループットアプリケーションに有効です(図2~4)。

図2. 非ホットスタートDNAポリメラーゼとホットスタートDNAポリメラーゼを用いたPCRの比較。 ホットスタートDNAポリメラーゼの使用により、目的増幅産物の収量が向上し、非特異的増幅が減少していることに注目してください。

図3. 室温での反応セットアップが可能なホットスタートDNAポリメラーゼはハイスループットアプリケーションに適している。 室温で調製したPCR反応液を、室温でそれぞれ0、24、72時間インキュベートした後、サーマルサイクラーにセットした。 室温でセットアップ後72時間が経過したサンプルであっても、ヒトgDNAを鋳型にした2 kb断片の特異的な増幅を確認できた。このことは、ホットスタートDNAポリメラーゼが、大規模実験に適していることを示している。
抗体の代わりに、酵素の活性部位を熱感受性化学修飾することにより酵素にホットスタート特性を持たせることができます。また、アプタマー等の低分子化合物の使用により活性化時間を短くすることも可能です。特異性を高めるためには、どのホットスタート技術を選択するかに関わらず、DNAポリメラーゼの活性を非加熱状態で効果的に阻害することが重要です(図4)。

図4. ポリメラーゼ活性の比較: (A)真の「ホットスタート」DNAポリメラーゼと(B)「ウォームスタート」DNAポリメラーゼ。 ポリメラーゼの活性は、60°C(一定)で60分間測定した。 熱活性化試験(青色カーブ)では、ポリメラーゼから抗体を分離するため、94°Cで2分間熱処理した。 熱活性化を行わない場合(赤色カーブ)、真のホットスタートDNAポリメラーゼでは活性が検出されなかったが、ウォームスタート酵素は60°Cで活性化が起こった。このことは、ウォームスタート酵素がホットスタートアプリケーションには適さないことを示している。
サーマルサイクルは、DNAを増幅する連鎖反応の繰り返しを可能とする主要な条件であるため、DNAポリメラーゼの耐熱性は、非常に重要です。Taq DNAポリメラーゼは、好熱性細菌由来で、比較的高い温度にも耐えることができますが、その半減期は90°C以上で著しく短くなります。このように耐熱性が不十分であるため、強固な二次構造やGCリッチ配列を持つDNAの増幅の際、高温長時間処理を行う必要があり、問題となりえます。 Taqポリメラーゼは、長鎖のテンプレートを増幅する際にも、より多くの、または追加のインキュベート時間を必要とする場合があります。このため、こうした問題を克服するため、より高い耐熱性を持つ超好熱性生物から分離されたDNAポリメラーゼが有用となってきました。
よく知られた超耐熱性酵素の1つであるPfu DNAポリメラーゼは、熱水環境で発見された古細菌、Pyrococcus furiosusから単離されました。Pfuポリメラーゼは、95°Cにおいて、Taqポリメラーゼの20倍も高い耐熱性を有しています[3]。その他の主な超耐熱性DNAポリメラーゼとしては、古細菌のThermococcus属およびPyrococcus属の一種から得られたKOD、GBDなどがあります。
古細菌のDNAポリメラーゼは、極めて耐熱性が高いものの、欠点もあります。例えば、超耐熱性Pfu DNAポリメラーゼは、その処理能力の低さ(Taq DNAポリメラーゼとの比較)ゆえに、DNAの合成速度が遅いという欠点があります。さらに、古細菌のDNAポリメラーゼには、DNA修復機構としてのウラシル結合ポケットが存在するため、ウラシルを含むDNAテンプレートを増幅することができません[4、5]。ウラシルを含むDNA配列は、バイサルファイト法を用いたメチル化解析、およびPCRキャリーオーバー防止に係るため、それらの用途には使用できません。
DNAポリメラーゼのプルーフリーディング活性は、DNA配列複製の正確性を向上させることで、そのフィデリティを決定付けます。ハイフィデリティDNAポリメラーゼは、強力なプルーフリーディング活性を持った酵素です。DNAポリメラーゼのDNA配列を正確に複製する性能(すなわち、誤りのない配列をもたらすこと)は、クローニング部位特異的変異導入などのアプリケーションにおいて重要となります。
DNAポリメラーゼのプルーフリーディング活性は、ヌクレオチドの誤取り込みを修正する3′→5′エキソヌクレアーゼ活性に基づいています。このエキソヌクレアーゼ活性部位は、DNAポリメラーゼ上の5′→3′ポリメラーゼ活性部位とは離れた位置に存在しています(図5)。ポリメラーゼドメインで間違ったヌクレオチドが取り込まれると、塩基対形成がうまくいかず、DNA合成が停滞します。この停滞により、DNAポリメラーゼによるミスマッチヌクレオチドの切り出し、および正しいヌクレオチドへの置換が可能となります[6]。

図5. 5′→3′ポリメラーゼ領域および3′→5′エキソヌクレアーゼ領域を持つDNAポリメラーゼ(図は、E. coli DNAポリメラーゼIの構造に基づく)。
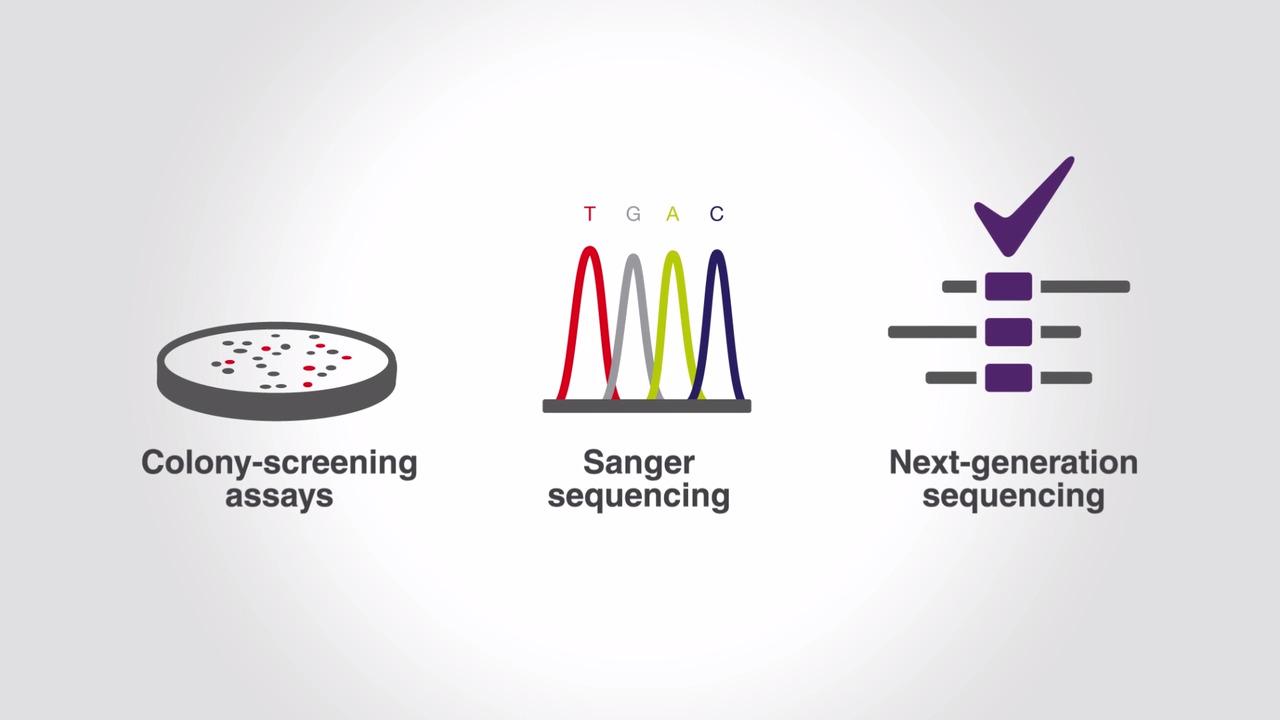
DNAポリメラーゼのフィデリティは、コロニースクリーニングアッセイ、サンガーシーケンシング、次世代シーケンシングなど、様々な方法で評価することができます[7~10]。昔からあるコロニースクリーニング法の場合、まずlac遺伝子のPCR増幅断片をプラスミドにクローニングします。組換えプラスミドで形質転換されたコロニーは、青色/白色コロニースクリーニングで評価することができます。突然変異したlac遺伝子インサート(PCR中の複製エラーによって導入されたと考えられる)を持つ場合は、LacZ活性を欠失しており、白色のコロニーが形成されます。一方、PCRエラーが起きなかったlacインサートを持つ場合は、青色のコロニーが形成されます。サンガーシーケンシングを用いた場合は、クローニングされたPCR断片のシーケンシングによって、DNAポリメラーゼのエラー率を特定することができます。次世代シーケンシングでは、PCR増幅産物を直接シーケンシングの対象とすることができます(図6)。

図6. DNAポリメラーゼのフィデリティを評価するための一般的な方法。
DNAポリメラーゼのフィデリティは、多くの場合、エラー率の逆数(フィデリティ = 1/エラー率)として表されます。エラー率は、ポリマー化したヌクレオチドの総数に対する間違って取り込まれたヌクレオチドの数を意味します。そのため、測定されたDNAポリメラーゼのフィデリティは、PCRアンプリコンの長さ、およびPCR産物の生成に用いたサイクル数に大きく依存します。様々なポリメラーゼのフィデリティを正確に比較するためには、同一の方法およびサイクルパラメーターを用いて測定を行う必要があります。
多くの場合、フィデリティは、Taq DNAポリメラーゼのフィデリティとの比較で表します。PfuおよびKODなど、天然のプルーフリーディングDNAポリメラーゼは、Taq のおよそ10倍のフィデリティを有しています。しかし、進化分子工学的手法を用いて開発された「次世代型」ハイフィデリティDNAポリメラーゼは、Taqポリメラーゼの50~100倍を超えるフィデリティを有しています(図7)。このような酵素を使用すると、間違った塩基が取り込まれる確率は、取り込まれたヌクレオチド全体の100万分の1になります。

図7. 次世代シーケンシング法による一般的なハイフィデリティ酵素の比較。
酵素の処理能力は、基質との結合1回あたりに処理されるヌクレオチドの数と定義されます。DNAポリメラーゼの処理能力は、多くの場合、合成速度だけでなくその基質への親和性も反映します。そのため、処理能力の高いDNAポリメラーゼは、長鎖テンプレート、二次構造を持つ配列やGCリッチ配列、およびPCR阻害物質(血液や植物の組織で認められるヘパリン、キシラン、フミン酸など)が存在する条件での増幅に有効です(図8)。

図8. 処理能力の高いDNAポリメラーゼは、(A)様々なサイズのヒトgDNAターゲット配列、(B)GCリッチ領域を含むテンプレート(エンハンサー不使用)を増幅可能で、また(C)一般的なPCR阻害物質を含むサンプルでも、標的配列を効果的に増幅することができる。 上記の実験には、処理能力が非常高い酵素である、Invitrogen™ Platinum™ SuperFi™ DNAポリメラーゼを使用した。
初期のハイフィデリティDNAポリメラーゼは、その強力なエキソヌクレアーゼ活性によってポリメラーゼ反応が減速され、処理能力が低い傾向にあります。そのため、長鎖DNAを標的とした場合、合成速度が著しく遅くなる場合があります。例えば、Pfu DNAポリメラーゼのフィデリティは、Taq DNAポリメラーゼの7倍にも及びますが、その合成速度は、Taq ポリメラーゼの半分以下です。別のタンパク質由来のDNA結合ドメインを付加したDNAポリメラーゼにより、処理能力が大きく向上しました(図9)[10]。このような改変DNAポリメラーゼの処理能力は、2~5倍に増強されています。

図9. DNAポリメラーゼの処理能力。 (A)高処理能力を持つDNAポリメラーゼは、その基質に対する親和性が高い傾向があり、結合1回あたりのヌクレオチド取り込み数が多い。 (B)改変DNA結合領域を持つDNAポリメラーゼ配列の概略図(Pol = ポリメラーゼドメイン、3-′5′ exo = 3′→5′エキソヌクレアーゼドメイン、DBD = DNA結合ドメイン、N = N末端、C = C末端)。
DNAポリメラーゼの4つの特性(特異性、耐熱性、フィデリティ、処理能力)を併せ持たせることで、汎用性が高い酵素を実現し、それらのPCRにおけるアプリケーションをより一層広めることができます。酵素の特異性によって、目的のPCR産物を高収量で確実に得るとともに、ダウンストリームアプリケーション(クローニング、定量など)における問題発生を最小限に抑えることができます。強力な処理能力および耐熱性によって、二次構造を持つ配列やGCリッチ配列、および長鎖DNAの増幅が容易になります。さらに、高度な処理能力により、サンプルのPCR阻害物質への耐性を獲得できます。最終的に、ハイフィデリティによって、正確な配列の複製を実現します。
参考文献
- Sharkey DJ, Scalice ER, Christy KG Jr (1994) Antibodies as thermolabile switches: high temperature triggering for the polymerase chain reaction. Biotechnology (N Y) 12(5):506–509.
- Kellogg DE, Rybalkin I, Chen S (1994) TaqStart Antibody: "hot start" PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase. Biotechniques 16(6):1134–1137.
- Pelt-Verkuil EV, Belkum AV, Hays JP (2008) Principles and Technical Aspects of PCR Amplification. Dordrecht: Springer.
- Lasken RS, Schuster DM, Rashtchian A (1996) Archaebacterial DNA polymerases tightly bind uracil-containing DNA. J Biol Chem 271(30): 17692–17696.
- Fogg MJ, Pearl LH, Connolly BA (2002) Structural basis for uracil recognition by archaeal family B DNA polymerases. Nat Struct Biol 9(12):922–927.
- Steitz TA (1999) DNA polymerases: structural diversity and common mechanisms. J Biol Chem 274(25):17395–17398.
- Lundberg KS, Shoemaker DD, Adams MW et al. (1991) High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 108(1):1–6.
- Barnes WM (1992) The fidelity of Taq polymerase catalyzing PCR is improved by an N-terminal deletion. Gene 112(1):29–35.
- Vandenbroucke 1, Van Marck H, Verhasselt P (2011) Minor variant detection in amplicons using 454 massive parallel pyrosequencing: experiences and considerations for successful applications. Biotechniques. 51(3):167–177.
- Kinde I, Wu J, Papadopoulos N et al. (2011) Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 108(23):9530–9535.
- Wang Y, Prosen DE, Mei L et al. (2004) A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res 32(3):1197–1207.
For Research Use Only. Not for use in diagnostic procedures.