Search Thermo Fisher Scientific
一般的な10のPCR法

先のセクションで述べたように、PCRは、いくつかの必要不可欠な反応構成要素を含む3つの主要なサーマルサイクリングステップから構成されます。以下のセクションで説明するように、用途に応じて収量の増加、特異性の向上またはアッセイ時間の短縮など特定の実験結果を達成するため、PCR設定の最適化をします(表1)。
表1.一般的なPCR方法およびそれらの中核となる利点
| PCR法 | 特異性 | 収量 | 時間 | 利便性 | 特殊アプリケーション |
|---|---|---|---|---|---|
| ホットスタートPCR | ✓ | ✓ | |||
| タッチダウンPCR | ✓ | ✓ | |||
| ネステッドPCR | ✓ | ✓ | |||
| 高速PCR | ✓ | ✓ | |||
| ダイレクトPCR | ✓ | ✓ | |||
| マルチプレックスPCR | ✓ | ✓ | |||
| 長鎖PCR | ✓ | ||||
| GCリッチPCR | ✓ | ||||
| インバースPCR | ✓ | ||||
| 定量PCR | ✓ |
関連ビデオ
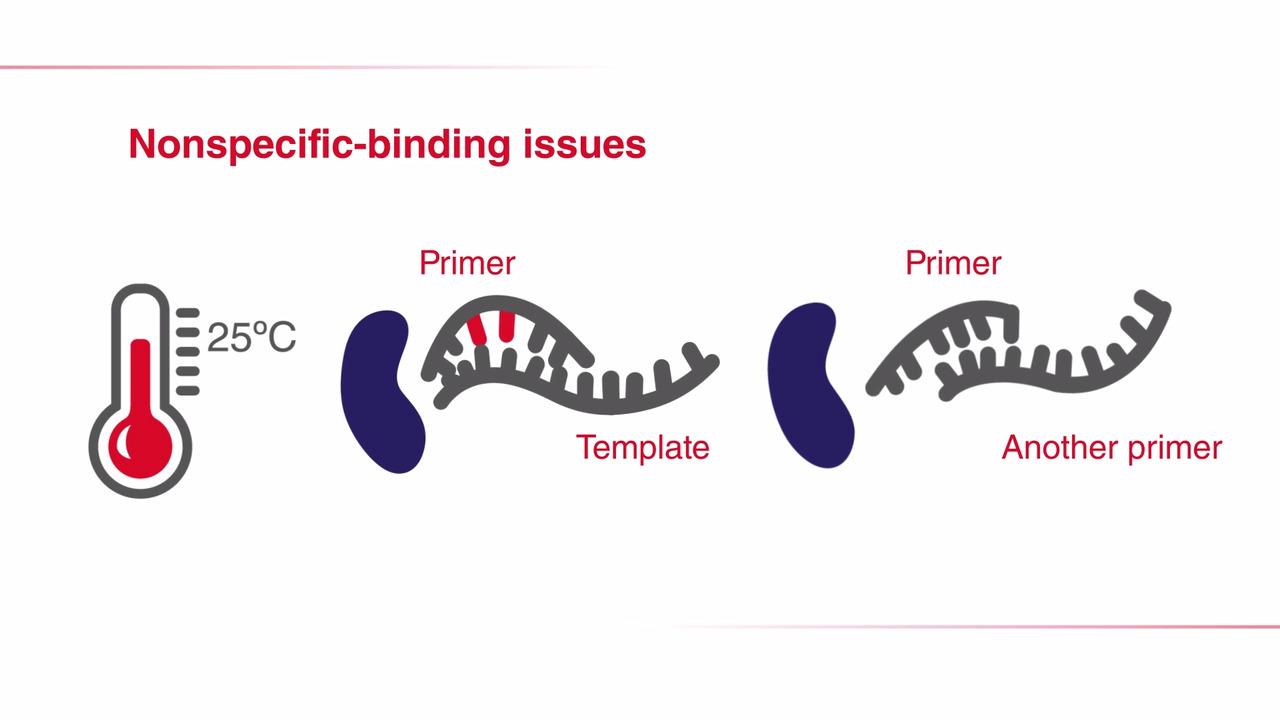
ホットスタートPCRは、通常、PCR増幅で特異性を高めるのに用います。ホットスタートPCR法は、抗体、アフィボディ、アプタマーといった酵素修飾因子、または室温でDNAポリメラーゼ活性を阻害する化学修飾を用います。これらの修飾は、相同性の低いテンプレート配列と結合したプライマー(ミスプライミング)、および互いに結合したプライマー(プライマーダイマー)に起因した非特異的な増幅を防止します。ホットスタート法ではDNAポリメラーゼの活性は室温で阻害されるため、特異性および増幅を大きく損なうことなく、高スループット実験の場合など、常温での多くの反応液調製を簡便にします(詳細はDNAポリメラーゼ特性を参照してください)。
反応液を調製した後、DNAポリメラーゼは最初の加熱ステップつまり「ホットスタート」で活性化され、このとき、酵素修飾因子が高温下(通常90°C以上)で遊離します(図1)。活性化時間および温度は、DNAポリメラーゼ、およびホットスタート修飾因子の性質に応じて変わります。いくつかのDNAポリメラーゼでは、活性化ステップと1サイクル目の変性ステップを1つに合わせてしまうこともあります。

図1. 抗体ベースのホットスタート技術に用いたDNAポリメラーゼ。
特異性を向上させる別のアプローチは、PCRサイクリングのパラメーターに変更を加えるというものです。タッチダウンPCRの場合、最初の数サイクルのアニーリング温度は、プライマーの最も高い融解温度(Tm)よりも数℃高く設定します[1,2]。より高い温度は、プライマーダイマーや非特異的プライマーテンプレート複合体の形成を不安定にするため、望ましくない増幅を最小限に抑えます。このように、より高いアニーリング温度は、PCRの開始時に非特異的なPCR産物を減らし、特異的な増幅を促進します(詳細はPCRアニーリングのステップを参照してください)。
より高いアニーリング温度は、プライマーダイマーや非特異的プライマーの結合を防止する一方で、目的とする標的からもプライマーが解離しやすくなるため、PCR収率の低下を招くことがあります。この課題を解決するため、最初の数サイクルの各サイクルでアニーリング温度を毎サイクル1°Cずつ下げることで、望ましいアンプリコンの十分な収量が得られます。アニーリング温度が最適温度(通常、最も低いプライマーTmよりも3~5°C低い)に到達(タッチダウン)したら、プライマーアニーリングの残りのサイクルの最後までその温度を維持します。このようにして、望ましいPCR産物を選択的に増加させると同時に、PCR反応の全体にわたり非特異的な標的がほとんどまたは全く増幅しないようにすることができます(図2)。

図2. タッチダウンPCR 本手法は、最適なアニーリング温度よりも高い温度で開始することで特異性(黄色の曲線)を促進します。続いて、サイクリングが継続するにつれて、温度はアニーリング最適温度に到達するまで徐々に低下します(黒色の直線)。 目的とするアンプリコン(緑色の曲線)は、最適化したアニーリング温度で十分な収量が得られます。
ネステッドPCRは、標準的なPCRの変法であり、望ましいアンプリコンの特異性および収率を高めます[3]。この手法では、2対のPCRプライマーをデザインします:1つめのセット(アウタープライマー)は、目的のアンプリコンを含むDNA領域の両側に位置するのに対し、2つめのセット(ネステッドプライマー)は、増幅したい目的のターゲット領域にデザインします。アウタープライマーは、PCRの初回ラウンドにおいて、隣接領域を含めて標的を増幅するのに使用します。次に、この初回ラウンドの産物は、ネステッドプライマーを用いる第2ラウンドのPCRでテンプレートとして用います(図3)。

図3. ネステッドPCR
プライマーの最初のセット(アウタープライマー)によるミスプライミングが原因で非特異的な産物が増幅された場合、第2のプライマーセット(ネステッドプライマー)によって同じ非特異的な領域が増幅される可能性は低いですが、アウタープライマーが目的の標的を認識した場合は、ネステッドプライマーによって特異性が向上します。2段階のPCRラウンドのもう一つの利点は、本アプローチにより、限られたDNAインプット量から望ましい標的の十分な収量を得やすくなることです。
高速PCRの場合、PCRステップの時間が短くなり、収量または効率に影響を与えることなく、増幅をより迅速に完了することができます。合成能力の高いDNAポリメラーゼは、各合成ステップの間により多くのヌクレオチドを取り込むことができるため、特に高速サイクリング条件に適しています(詳細は合成能力を参照してください)。合成能力の高いDNAポリメラーゼは、合成能力の低いTaqポリメラーゼに必要な伸長時間の1/2~1/3に相当するPCR伸長時間で、高い増幅効率を得ることができます(図4)。プライマーのアニーリングステップと伸長ステップの温度が互いに数度以内である場合、両者を合わせてひとつのステップにすることで、PCR時間をさらに短縮することができます。本手法は、2ステップPCRプロトコルという名前でも知られています。

図4. ヒトgDNAから得られた3.8 kb断片の増幅での、低い合成能力と高い合成能力のDNAポリメラーゼの比較。 2ステップPCRプロトコル(アニーリングと伸張ステップを合わせて1つにした)を実施しました。
Taqポリメラーゼのような低い合成能力のDNAポリメラーゼを使用する場合、標的が500 bp未満の短鎖なら、高速サイクリング条件を適用できる可能性があります。このサイズのアンプリコンは、一般に長い重合時間を必要としないため、PCRプロトコルの伸張ステップを短縮できる可能性があります。収量を低下させない最短の伸長時間を見つけるため、伸長時間を段階的に短縮(秒単位で)させるという方法でPCRを実施することもあります。各標的およびプライマーセットは、結果が変動しやすい傾向があるため、特定の条件下で高速PCRの最適化をする必要があります。
高速PCRのもう一つの改変は、変性時間を短縮する一方、その埋め合わせとして温度を98°Cに上げるというものです。この方法で注意を要するのは、あまり熱に安定でない酵素はそのような高温で容易に変性してしまうということです(詳細はDNAポリメラーゼの熱安定性を参照してください)。
表2. 合成能力の低いDNAポリメラーゼを用いて高速PCRを達成するための反応パラメーター。
| パラメーター | 高速PCRの最適化 |
|---|---|
| アンプリコンの鎖長 | < 500 bp |
| 変性時間 | 減少 |
| 変性温度 | 増加 |
| 伸長時間 | 減少 |
サイクリング時間が高速のサーマルサイクラー、および薄壁のPCRプラスチック製品を用いれば、それぞれ高速の温度移行速度および効率的な熱伝達により、PCRが非常に速くなります。
ダイレクトPCRとは、核酸を分離することなく、直接試料から標的DNAを増幅することを指します。ダイレクトPCRで、細胞または組織などの試料は、特別に調製された緩衝液中で溶解し、高温変性ステップ中にDNAを遊離します。そのため、この方法はワークフローを簡素化し、操作時間を節約し、精製ステップが原因のDNA損失を防止します(図5)。

図5. 従来型PCR対ダイレクトPCR。
合成能力の高いDNAポリメラーゼは、しばしば直接PCR用に推奨されます。細胞の破片、タンパク質、脂質および多糖類がDNAとともにライセート中に遊離し、PCRを阻害します。合成能力の高いDNAポリメラーゼは、そのような阻害物質に耐性があり、ダイレクトPCRの実施を可能にします。合成能力の高い酵素は、より高感度であることが多く、未精製の試料から少量のDNAを効率よく増幅するのに用いることができます。
GC含量の高い(65%超)DNAテンプレートは、G塩基とC塩基との間のより強い水素結合のため、増幅するのが困難になります。GCに富む配列は、2次構造にも関係しています。それゆえ、GCに富む配列は、DNAポリメラーゼをテンプレートに沿って「つっかえつっかえ進ませる」ため、DNA合成を妨げます。
GCに富む標的を増幅するには、プライマーが結合してDNAポリメラーゼが合成できるよう、2本鎖テンプレートを1本鎖に解離させなければなりません。強力なGC相互作用に打ち勝つために最もよくみられるアプローチは、DNAを変性しやすくするDMSOのようなPCR添加剤または補助溶剤を利用するという方法です(図6A)。こうした試薬は、プライマーTmを下げることが多いため、アニーリング温度を適宜調整する必要があります。
合成能力の高いDNAポリメラーゼは、プライマー伸長中にテンプレートと強力に結合するため、GCリッチPCRに有利です(図6B)。より高い変性温度(例:95°Cの代わりに98°C)は二本鎖の解離およびPCR増幅を促進する可能性があるため、非常に熱に安定なDNAポリメラーゼもGCリッチPCRに有用です(詳細はPCRサイクリングを参照してください)。

図6. 様々なGC含量を有するヒトgDNA領域の増幅。(A) GC含量76%の約0.8 kbの標的を、合成能力の低いDNAポリメラーゼを用いて増幅しました。 特異性を向上させるため、DMSOの添加量を増加させました。 (B) 様々なGC含量を有する7つの断片を、合成能力の高いDNAポリメラーゼを用いて増幅しました。 GC含量70%および76%の断片に対してのみ、GCエンハンサーを使用しました。
マルチプレックスPCRは、単一のPCRチューブ中で、異なる標的の同時並行的な増幅を可能にします。マルチプレキシングは、時間、試薬および試料を節約するのみならず、多くのアンプリコンの同時比較も可能にします(図7)。

図7. シングルプレックスPCR vs. マルチプレックスPCR シングルプレックスPCRの各反応は、1つの標的を増幅するための1つのプライマーセットを含みます。 マルチプレックスPCRでは、1つの反応で多くの標的を増幅するため、多くのプライマーセットを使用します。
マルチプレックスPCRの場合のように多くのプライマーセットが単一チューブ内に存在する場合、反応を単一のプライマーセットおよび標的に対して最適化することはできず、すべてのプライマーおよび標的全体に対して最適化することになるため、非特異的な増幅および効率の低下が懸念されます。そのため、非特異的な増幅につながるようなミスプライミングを最小限に抑えるプライマーデザインが極めて重要となります。プライマー配列は、それらの標的にできるだけ特有でなければならず、また、すべてのプライマーのTmは、互いに5°C以内でなければなりません。マルチプレキシングの実施前に、各プライマーセットの特異性および効率を、シングルプレックス反応の中で検証しなければなりません。さらに、アンプリコンは、ゲル電気泳動で分離して識別できる異なったサイズでなければなりません。プライマーデザインとアンプリコンサイズに加え、ホットスタートDNAポリメラーゼおよびマルチプレキシング用に特別に調製したPCR緩衝液は、PCRの成功および特異性を向上させることができます(図8)。

図8. ゲル電気泳動で示されたシングルプレックスおよびマルチプレックスPCR結果。Invitrogen™ Platinum™ Multiplex PCR Master Mix を本実験に使用しました。
マルチプレックスPCRは、エンドポイント反応として日常的に行われていますが、標的増幅物のマルチラベリングおよび検出の能力を備えたリアルタイムPCRによるアプローチの方が高い定評があります。マルチプレックスリアルタイムPCRも、ヒトの識別を目的とした遺伝子マーカーの検出にしばしば利用されます。
長鎖PCRとは、一般に、5 kbを超えるDNA標的を増幅することを指します。長鎖PCRは、古典的にTaq DNAポリメラーゼ(高速伸長)とハイフィデリティ酵素(正確性)の混合物を用いて実施されます。
合成能力の高いハイフィデリティDNAポリメラーゼが発明されたため、現在の長鎖PCRは、正確性が大幅に改善されており、以前よりも短い時間で実施することができます。高い合成能力は、長い断片(例:gDNAから得られた20 kb超)を数時間で増幅できる強力なDNA結合ドメインをデザインすることにより達成されます(図9、表3)。さらに、非常に高いフィデリティ(例:Taqポリメラーゼに比べて100倍超のフィデリティ)があるため、長い断片の複製のエラー率を確実に低く抑えることができます(表3)。(詳細はDNAポリメラーゼの特性を参照してください。)

図9. 合成能力の高いDNAポリメラーゼを用いた長い断片の増幅。 15 kbおよび30 kb断片の特異的な増幅物がヒトgDNA試料から得られました。
表3.長鎖PCRおよびクローニングにおける、合成能力の高い、遺伝子操作で作り変えたハイフィデリティDNAポリメラーゼの利点。
DNAポリメラーゼの合成能力が高いため、長鎖PCRの反応時間が有意に短縮(この例では半減)される一方で、フィデリティが高いため、エラーのないインサートをもつコロニーを得るためのスクリーニングに要する労力が減ります。
| Taq DNAポリメラーゼとプルーフリーディングDNAポリメラーゼの混合物 | 合成能力の高い、遺伝子操作で作り変えたハイフィデリティDNAポリメラーゼ | |
|---|---|---|
| 伸長速度 | 60 sec/kb | 30 sec/kb |
| PCR時間 (20 kbの標的、30サイクル) | 約10.5時間 | 約5.2時間 |
| フィデリティ (Taq DNAポリメラーゼに比べて) | 5倍 | 100倍超 |
| クローニングのエラー率 (20 kb、30サイクルのPCR) | 1クローン当たり平均2.5エラー | 4つのうち1つのクローンがエラーを含む可能性あり |
10 kb超の標的を増幅する場合、PCRプロトコルは、以下の5つの重要な領域で最適化が必要になる可能性があります:
- DNA試料が良好な品質および純度を確実に有するようにすること。
- 熱安定性が低い場合、活性の損失を補うため、サイクリング時間を延ばしてより多くの量のDNAポリメラーゼを使用すること。
- プライマー結合を促進するため、アニーリングステップおよび伸張ステップの温度を下げること。
- テンプレートDNAの完全な1本鎖への解離およびプライマーの結合を促進するため、PCRステップの時間を延ばすこと。
- 標的領域の完全長複製を確実にするため、PCR伸長時間を適宜延長すること。
インバースPCRは、当初、隣接する未知領域の配列を決定するためにデザインされました。インバースPCRは、遺伝子のプロモーター配列を調べるのに有用です:遺伝子融合、転座および転位、ならびにウイルス遺伝子組込みといった発がん性染色体再配列。本手法は、プライマーが普通のPCRの場合のように互いに向かい合う向きでなく、互いに離れる向きに伸長するようにデザインされるため、インバースPCRとして知られています[4,5]。今日、インバースPCRは、部位特異的変異導入に日常的に利用され、標的プラスミドを複製する一方で、望ましい変異を導入します。
ゲノムDNAの未知配列を調べる従来型ワークフローでは、制限酵素処理およびライゲーションの後にインバースPCRを実施し、次いでPCRアンプリコンのシーケンシングと続きます。gDNA断片化では、制限酵素を選択することで適切な長さの断片を生成し、セルフライゲーションします。また、制限酵素は既知の配列を切断しないものを選択することにより、ライゲーションは隣接する未知配列の間で起こります。ライゲーションは、低濃度の制限酵素処理されたDNA断片を用いることで、セルフライゲーションがマルチ断片ライゲーション(すなわち、鎖状体形成)よりも起こりやすいように最適化します。
セルフライゲーション後、DNAの既知の領域からプライミングすることにより、インバースPCRを実施します。その結果得られたアンプリコンは、各末端に既知のDNA配列の一部を含んでいます。これらのアンプリコンは、末端からシーケンシングして、既知配列に隣接する未知の領域を調べることができます(図10)。

図10. 隣接する未知配列の増幅および特性評価を目的としたインバースPCR。
配列の増幅量の程度(収率)はテンプレートのインプット量に左右されるため、一般に、PCRは試料中に存在するDNAの量を定量するのに利用されており、その最も一般的な用途は遺伝子発現の定量です。エンドポイントPCRは、可能な手法の1つですが、検出感度の限られるゲル電気泳動で収率を測定しなくてはならないという大きな欠点があります。さらに重要なことに、増幅がプラトーに到達したPCRの終端に定量が行われた場合(図11)、DNAゲル染色の強度は、DNAインプット量と直線的に相関しません。エンドポイントPCRでプラトーに到達する前に遺伝子発現を半定量分析する場合、段階希釈したDNA試料をインプットサンプルとして使用するか、またはアンプリコンを特定のPCRサイクルで回収することで、ゲルのシグナル強度により遺伝子発現量を推定できます[6,7]。

図11. PCRの増幅曲線または反応力学。 エンドポイントPCRの場合、アンプリコンはPCR後に増幅がプラトーに到達したときに検出されます。 リアルタイムPCRの場合、アンプリコンは対数増幅期に定量されます。
エンドポイントPCRをベースとした定量の限界は、Higuchiらによる蛍光シグナルを用いたPCR増幅のリアルタイムモニタリングの報告により克服されました[8]。本手法は、現在の定量PCR(qPCR)のベースになりました。1997年に最初のqPCR機器が市販されたことで、遺伝子発現量およびコピー数を正確に定量するPCRの検出が可能となりました[9,10]。qPCRは、対数増幅期での標的増幅に伴う蛍光強度のリアルタイムモニタリングに基づいており(図11)、エンドポイントPCR定量の問題点を回避しています。qPCRは、相対的および絶対的な遺伝子発現量を定量的に測定するとはいえ、その定量は、検出能力に起因してまだ限定的です。
DNA試料の真の絶対定量は、1990年代にリアルタイムPCRと並行して開発された手法であるデジタルPCR(限界希釈PCRとも呼ばれる)を用いて可能になりました[11-13]。デジタルPCRでは、高度に希釈したDNA試料がマルチコンパートメントチップの中に分配され、各区画は目的とする標的を1コピー以下しか含みません。各区画内の増幅を測定することで、陽性または陰性の結果が得られます(それぞれ1または0コピーという「デジタル」の結果)。試料のコピー数は、陰性反応の分画から統計モデル(ポアソン分布)を用いて決定します。定量に既知の試料(基準)は必要ありません(図12)。遺伝子発現量およびコピー数の測定に加え、デジタルPCRは、低頻度アレルの識別、ウイルス力価測定、および次世代シーケンシングライブラリーの絶対定量といった用途に適しています。

図12. 絶対的定量を目的としたデジタルPCRの一般的なワークフロー。
要約すると、増幅結果を改善するため、変更を加えたPCRプロトコルおよびDNAポリメラーゼが日常的に利用されています。PCRの基本的概念は変わらないままですが、PCRの新しい手法は、分子生物学の研究を進歩・効率化させ続けています。
参考文献
- Don RH, Cox PT, Wainwright BJ (1991) 'Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res 19(14):4008.
- Hecker KH, Roux KH (1996) High and low annealing temperatures increase both specificity and yield in touchdown and stepdown PCR. Biotechniques 20(3):478–485.
- Haff LA (1994) Improved quantitative PCR using nested primers. PCR Methods Appl 3(6):332–337.
- Ochman H, Gerber AS, Hartl DL (1988) Genetic applications of an inverse polymerase chain reaction. Genetics 120(3):621–623.
- Pavlopoulos A (2011) Identification of DNA sequences that flank a known region by inverse PCR. Methods Mol Biol 772:267–275.
- Raeymaekers L (1999) General Principles of Quantitative PCR. In: Kochanowski B, Reischl U (editors), Quantitative PCR Protocols. Totowa, NJ: Humana Press. pp. 31–41.
- Siebert PD (1999) Quantitative RT-PCR. In: Kochanowski B, Reischl U (editors), Quantitative PCR Protocols. Totowa, NJ: Humana Press. pp. 61–85.
- Higuchi R, Fockler C, Dollinger G (1993) Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y) 11(9):1026–1030.
- Wittwer CT, Ririe KM, Andrew RV (1997) The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques 22(1):176–181.
- Applied Biosystems, Inc. (1997) Relative quantitation of gene expression: ABI PRISM 7700 Sequence detection system. User Bulletin #2: Rev B part 4304859B, 1–36.
- Sykes PJ, Neoh SH, Brisco MJ et al. (1992) Quantitation of targets for PCR by use of limiting dilution. Biotechniques 13(3):444–449.
- Kalinina O, Lebedeva I, Brown J et al. (1997) Nanoliter scale PCR with TaqMan detection. Nucleic Acids Res 25(10):1999–2004.
- Vogelstein B, Kinzler KW (1999) Digital PCR. Proc Natl Acad Sci U S A 96(16):9236–9241.
For Research Use Only. Not for use in diagnostic procedures.