Search Thermo Fisher Scientific
Compound Discovererソフトウェア

サンプルから構造まで;知見取得にかかる時間を短縮
データセットの大小にかかわらず、フルスキャンおよびMSnで得られた低分子データから既知の化合物および分子経路を同定します。Thermo Scientific Compound Discovererソフトウェアを導入すれば、親化合物が既知である未知化合物向けの高度なソフトウェアツールがすべてそろいます。Compound Discovererソフトウェアを使用すれば、価値あるデータから迅速に知見を得るための強力な統合型ソフトウェアワークフローにより、化合物同定を効率化し、サンプル間の実際の差を測定して生物学的経路を解明できます。
Compound Discovererは、メタボロミクスからバイオマーカーの発見、環境および食品の安全性、医薬品の代謝物または不純物の同定、抽出物および浸出物、法医学または臨床毒性学まで、どのような低分子研究アプリケーションにも対応する、他にはない低分子構造同定ソフトウェアです。
クイックリンク
Compound Discovererソフトウェアの主な利点
| クリック回数を削減 | 未知化合物を特定 | サンプルセット間の真の差異を検出 | 生物学的パスウェイを解明 |
|---|---|---|---|
| カスタムワークフロー、柔軟な視覚化およびグループ分けツールによりデータ解析および処理を制御できます。カスタマイズ可能な報告方法によって結果を共有するかまたは Thermo Scientific TraceFinderソフトウェアに直接結果を転送して、的を絞った解析を行えます。 | オンラインのmzCloudスペクトルライブラリ、ラボラトリー内のThermo Scientific mzVaultスペクトルライブラリおよび多数のサードパーティーの情報源を検索するマススペクトルライブラリを用いて、未知化合物を迅速かつ高い信頼性で同定できます。 | サンプルセット間の真の統計的差異をすばやく検出します。インタラクティブにリンクした画面を使用して、研究全体の成分の傾向を確認したり、複数のサンプルグループ間で興味深い重要な成分を同定したりすることが可能です。 | 代謝フラックス実験の実施、Thermo Scientific Metabolika、KEGG、BioCycデータベースを使用した経路の表示、検出化合物およびフラックス情報の経路への直接マッピングなどを行えます。 |

生体がどのように医薬品と相互作用するかの解明、ペプチドの環境動態の追跡、新たな化合物の開発、ブランドの評判の維持、基礎研究の実施など、目的がどのようなものであれ、低分子を徹底的に特性評価し、同定することは重要です。当社のeBookをダウンロードして、詳細をご覧ください。
Compound Discovererソフトウェアでワークフローを作成
Compound Discovererソフトウェアは、大規模かつ柔軟でカスタマイズ可能なデータ処理用ツールキットです。既定のワークフローテンプレートが用意されているため、すぐに立ち上げて使用できます。または、ご自身の実験用にデザインしたワークフローにテンプレートをすぐ適用できます。
ワークフローエディタおよび処理の手順

Click to enlarge
The workflow editor is driven by templates that can be assembled into a workflow using drag-and-drop functionality. Your custom workflow then enables comprehensive data processing and visualization based upon your experimental needs.
The workflow editor is driven by templates that can be assembled into a workflow using drag-and-drop functionality. Your custom workflow then enables comprehensive data processing and visualization based upon your experimental needs.
Study manager

Click to enlarge
Study manager uses a wizard to guide you through the process of setting up a study, including defining sample types and other variables such as multiple time points or sample variants, such as different yeast strains.
Study manager uses a wizard to guide you through the process of setting up a study, including defining sample types and other variables such as multiple time points or sample variants, such as different yeast strains.
解結果

Click to enlarge
The Results analysis interactively links your data and allows you to choose how to visualize it. When selecting samples or components, each view is automatically updated based upon your selection(s).
The Results analysis interactively links your data and allows you to choose how to visualize it. When selecting samples or components, each view is automatically updated based upon your selection(s).
アノテーションおよび生物学的解釈

Click to enlarge
Whether it is for unknown identification and spectral annotation or flux analysis and interpretation and mapping of biological information, Compound Discoverer software allows you to easily report your results, store your data and share your insights.
Whether it is for unknown identification and spectral annotation or flux analysis and interpretation and mapping of biological information, Compound Discoverer software allows you to easily report your results, store your data and share your insights.
Compound Discovererソフトウェアは、正確で一貫した高分解能データが得られるThermo Scientific Orbitrapベースの質量分析計の恩恵を受けています。このデータを活用して、本ソフトウェアはサンプル間でコンポーネントをそろえて、元素組成を決定し、ライブラリとマッチングして未知化合物を同定します。

Click to enlarge
The consistent mass accuracy and high-resolution spectral data from Orbitrap-based MS systems enables fine isotopic information to be obtained, as shown above for the compound davunavir. The resolution and accuracy provide confidence in elemental composition assignments and subsequent library matching, which can be further confirmed using MS/MS fragmentation information.
The consistent mass accuracy and high-resolution spectral data from Orbitrap-based MS systems enables fine isotopic information to be obtained, as shown above for the compound davunavir. The resolution and accuracy provide confidence in elemental composition assignments and subsequent library matching, which can be further confirmed using MS/MS fragmentation information.
結果を解釈して知見を獲得
簡易的な研究でも大規模研究でも、豊富な情報を含む複合的なデータが得られます。データから得られた知見を最大限活用するために、Compound Discovererソフトウェアは慎重にデータ処理を行った後、知見に富むデータを確認して紐付けることができます。
お客様が行うのが単一サンプルの分析でも大規模な大容量サンプル研究でも、Compound Discovererソフトウェアなら、以下のように未知低分子のデータ処理に必要なものがすべてそろっています:
- 未知ピークの検出
- 高度な統計ツール
- インタラクティブなデータ視覚化機能
- 化合物アノテーションツール
- 統合型データベースおよびマススペクトルライブラリ
- 生化学経路マッピング
- 安定同位体標識による非標的解析
- 大規模研究用の標準化ツール
研究のサンプル数にかかわらず、各サンプルには豊富な生データポイントがあります。こうしたデータポイントの一部は相互に関係していますが、多くは無関係です。複雑ではあるものの高品質なMSおよびMS/MS情報を解釈するには、データを絞り込んで意味のあるデータにたどり着く必要があります。

Click to enlarge
Within each data file there can be many millions of individual data points depending upon the relative complexity of the sample. To confidently obtain results for each sample, and across samples, each data file must have its array of data points aligned with features detected, e.g., a single compound with multiple isotopic peaks may also have numerous adducts. Once the data has been reduced from raw data into features, components can be assembled and identified.
Within each data file there can be many millions of individual data points depending upon the relative complexity of the sample. To confidently obtain results for each sample, and across samples, each data file must have its array of data points aligned with features detected, e.g., a single compound with multiple isotopic peaks may also have numerous adducts. Once the data has been reduced from raw data into features, components can be assembled and identified.
アプリケーションごとに作成された複数のテンプレートの一つを使用するか、それらのテンプレートの一つを編集することにより、ドラッグ&ドロップでワークフローを設定でき、データ処理を迅速かつ簡単に行えます。各処理ステップは、ワークフローツリー内のノードとして表されます。ノードをつなげることで、研究の要件に基づくデータ処理や解釈を進めることができます;ソフトウェア開発キットを使用するか、またはRやPythonのカスタムスクリプトを用いてノードを作成可能で、作成後のノードはScripting Nodeと共に使用して目的に合わせたワークフローを作成できます。

Click to enlarge
An example of a pre-defined workflow template for untargeted metabolomics. This template is designed to find and identify differences between samples. Each node is linked and performs a specific task. Here, retention time alignment is performed before unknown compounds are detected and grouped across all samples within the study. Elemental compositions are predicted using the accurate mass data, with compounds identified using the mzCloud mass spectral library and MS/MS information. Where there is no match from mzCloud, ChemSpider is used. For results with a ChemSpider match, mzLogic is used to rank results by likelihood of a match. Resulting compounds are then mapped to biological pathways using Metabolika. If QC samples are present, then normalization is performed, and subsequent differential analysis calculated (t-test or ANOVA).
An example of a pre-defined workflow template for untargeted metabolomics. This template is designed to find and identify differences between samples. Each node is linked and performs a specific task. Here, retention time alignment is performed before unknown compounds are detected and grouped across all samples within the study. Elemental compositions are predicted using the accurate mass data, with compounds identified using the mzCloud mass spectral library and MS/MS information. Where there is no match from mzCloud, ChemSpider is used. For results with a ChemSpider match, mzLogic is used to rank results by likelihood of a match. Resulting compounds are then mapped to biological pathways using Metabolika. If QC samples are present, then normalization is performed, and subsequent differential analysis calculated (t-test or ANOVA).
Compound Discovererソフトウェアのインターフェースは、投げかけられた質問ともっとも関連性の高い情報を表示するので、結果をスムーズに確認できます;各プロットと表はリンクしているため、確認中の化合物またはサンプルを反映して表示がすぐに更新されます。

Click to enlarge
The compound results table default layout where the chromatographic overlay (top left) shows the extracted ion chromatograms for each related adduct, as shown in the selected compound spectra (top right). The results table can be tailored to display the information relevant to your study, such as compound annotation, retention time, peak areas, statistical information, MS2 spectral match, and more.
The compound results table default layout where the chromatographic overlay (top left) shows the extracted ion chromatograms for each related adduct, as shown in the selected compound spectra (top right). The results table can be tailored to display the information relevant to your study, such as compound annotation, retention time, peak areas, statistical information, MS2 spectral match, and more.

Click to enlarge
Compound Discoverer software allows you to open related tables to quickly access the information used to generate annotations. This figure shows the information related to searching the mzCloud mass spectral library (bottom) along with the related experimental and library fragmentation spectra displayed as a mirror plot (top right).
Compound Discoverer software allows you to open related tables to quickly access the information used to generate annotations. This figure shows the information related to searching the mzCloud mass spectral library (bottom) along with the related experimental and library fragmentation spectra displayed as a mirror plot (top right).

Click to enlarge
From volcano plots from differential analysis (left), S-Plots from partial least squares discriminant analysis (middle), and hierarchical clustering analysis (right), it is easy to visualize complex data sets and determine what is statistically different using Compound Discoverer software. Each plot is active, so data points selected in the plot can be marked in the results tables and vice versa, helping determine the cause of observed differences or similarities and tracking compounds in complex data sets.
From volcano plots from differential analysis (left), S-Plots from partial least squares discriminant analysis (middle), and hierarchical clustering analysis (right), it is easy to visualize complex data sets and determine what is statistically different using Compound Discoverer software. Each plot is active, so data points selected in the plot can be marked in the results tables and vice versa, helping determine the cause of observed differences or similarities and tracking compounds in complex data sets.
Compound Discovererソフトウェアのアプリケーション
Compound Discovererソフトウェアはメタボロミクスから環境および食品の安全性、医薬品開発、法中毒学まで、さまざまなアプリケーションでご使用になれます。
代謝研究は非常に複雑なため、高品質かつ網羅的なデータを確実に取得することは、そのデータを分析して知見を得ることと同様に困難です。完全なサンプルカバレッジを実現するには通常、データ依存的取得法(DDA)のためのインクルージョンリストおよびエクスクルージョンリストの作成に多くの手作業を要します。
AcquireXの自動ワークフローでは、バックグラウンドイオンの自動除去および真のサンプル構成因子にフォーカスしたデータ取得を行う高度なMS/MSサンプリングにより、すべてのサンプル構成因子を直接照合できます。

Click to enlarge
AcquireX generates an exclusion list from a blank run (matrix matched). Then, an injection of the sample, followed by feature detection and component assembly, populates the inclusion list with compounds detected in the samples. A series of iterative DDA injections follow. Each injection is informed from the previous one, minimizing redundant fragmentation spectra and maximizing relevant spectra and metabolite annotations.
AcquireX generates an exclusion list from a blank run (matrix matched). Then, an injection of the sample, followed by feature detection and component assembly, populates the inclusion list with compounds detected in the samples. A series of iterative DDA injections follow. Each injection is informed from the previous one, minimizing redundant fragmentation spectra and maximizing relevant spectra and metabolite annotations.

Click to enlarge
Using DDA with AcquireX significantly increases the number of unique compounds with high-quality fragmentation spectra, so you obtain a more comprehensive picture of what is in your samples, as well as increasing the depth of decision-making MS/MS information available.
Using DDA with AcquireX significantly increases the number of unique compounds with high-quality fragmentation spectra, so you obtain a more comprehensive picture of what is in your samples, as well as increasing the depth of decision-making MS/MS information available.
AcquireXを他のCompound Discovererソフトウェア活用ツールと合わせて使用すれば、MS/MSスペクトルが得られない化合物の数を劇的に削減でき、高い信頼性での同定および、ランク付けによる推定的同定ができる化合物の数を大幅に増加させることができます。

Click to enlarge
Using DDA with AcquireX improves data quality and creates a significant increase in the number of compounds with MS/MS spectra, resulting in improved mzLogic ranking and higher mzCloud similarity scores, ultimately providing higher overall confidence in compound identification and putative unknown identification.
Using DDA with AcquireX improves data quality and creates a significant increase in the number of compounds with MS/MS spectra, resulting in improved mzLogic ranking and higher mzCloud similarity scores, ultimately providing higher overall confidence in compound identification and putative unknown identification.
安定同位体標識が非標的メタボロミクス研究に役立つ場合もあり、Compound Discovererソフトウェアはこうしたワークフローに対応するさまざまなデータ確認ツールおよび視覚化ツールを備えています。Compound Discovererソフトウェアは、リファレンスファイルで認められた非標識化合物の化学式に基づき、標識された化合物(アイソトポログ)を自動的に検出します。処理後、置換率(取り込み率)をプロットして複数ファイルで応答を確認したりMetabolikaの経路データ上に重ね合わせたりできます。

Click to enlarge
Stable isotope labelling uses the high-resolution mass spectral data from Orbitrap-based MS, where isotopologues can easily be detected and the respective elemental compositions determined. Compound Discoverer software makes it easy to visualize the amount of label incorporation and resulting isotopic distribution with the ability to map powerful qualitative and quantitative flux analysis information directly onto biological pathways in Metabolika.
Stable isotope labelling uses the high-resolution mass spectral data from Orbitrap-based MS, where isotopologues can easily be detected and the respective elemental compositions determined. Compound Discoverer software makes it easy to visualize the amount of label incorporation and resulting isotopic distribution with the ability to map powerful qualitative and quantitative flux analysis information directly onto biological pathways in Metabolika.
統計解析およびデータ標準化セクションに記載のとおり、Compound Discovererソフトウェアを使用すると、幅広い範囲で単変量および多変量解析が行えます。
Compound Discovererソフトウェアは脱アルキル化/ジアリル化および代謝全般を構造的に予測するインテリジェントな機能を備えているため、関心のある代謝物を見つけ、同定し、報告することができます。不純物および分解生成物の同定も同様のワークフローで行いますが、さまざまなソフトウェアツールおよびカスタマイズ可能なアプローチにより、複合的なサンプル中の関連する構成因子を高い信頼性で検出できます。
フラグメンテーションスペクトルの構造アノテーションに使用するフラグメントイオンサーチ(FISh)により、未知化合物の解明に加え、トランスフォーメーションする可能性がある位置の特定も行えます。

Click to enlarge
The FISh scoring node enables fragment structure annotation and uses the Thermo Scientific HighChem Fragmentation Library for real data from more than 52,000 fragmentation schemes to help localize (bio)transformation. Exact matches are shown in green, with transformation-shifted matches highlighted in blue (above), showing how the site of transformation is identified.
The FISh scoring node enables fragment structure annotation and uses the Thermo Scientific HighChem Fragmentation Library for real data from more than 52,000 fragmentation schemes to help localize (bio)transformation. Exact matches are shown in green, with transformation-shifted matches highlighted in blue (above), showing how the site of transformation is identified.

Click to enlarge
Pattern scoring allows you to flag compounds that match user-specified natural or artificial isotopic patterns. Including and using additional traces such as UV, PDA, CAD and analog, such as radio label traces, as shown above, ensures that minimal potential metabolites, impurities or degradants are missed.
Pattern scoring allows you to flag compounds that match user-specified natural or artificial isotopic patterns. Including and using additional traces such as UV, PDA, CAD and analog, such as radio label traces, as shown above, ensures that minimal potential metabolites, impurities or degradants are missed.
化合物クラススコアリングノードは、見落としがないようにするためのもう一つのツールです。一つの化合物クラス中の1つ以上の既知分子から得られた代表的なフラグメントのセットを使用して、関連性のあるまたは同じクラスの他の化合物を同定します。

Click to enlarge
The Compound Class scoring node allows you to identify compounds that are structurally related, ensuring that nothing is missed, from metabolites to potentially toxic or harmful extractables, leachables or degradants.
The Compound Class scoring node allows you to identify compounds that are structurally related, ensuring that nothing is missed, from metabolites to potentially toxic or harmful extractables, leachables or degradants.
Compound Discovererソフトウェアは、マトリックス干渉を低減するとともに、関連するマスディフェクトに基づいて特定の化合物クラスに標的を絞ることによってサンプルの複雑性を低下させるため、複合的なデータセットの同定、検出および確認をより迅速に行えます。

Click to enlarge
Upper left shows the Total Ion Chromatogram (TIC) for a sample in bile matrix, illustrating the potential complexity and matrix interferences present; bottom left shows the resulting trace following the use of Multiple Mass Defect Filtering (MMDF) and how it can be used to effectively simplify complex matrix samples such as bile, feces, blood, and plasma. The plot on the right demonstrates how the mass defect plot can be used to visualize data and mine using Kendrick formulas, for example unknown polymer identification. All data is interactively linked between plots and data tables within Compound Discoverer software to streamline data review.
Upper left shows the Total Ion Chromatogram (TIC) for a sample in bile matrix, illustrating the potential complexity and matrix interferences present; bottom left shows the resulting trace following the use of Multiple Mass Defect Filtering (MMDF) and how it can be used to effectively simplify complex matrix samples such as bile, feces, blood, and plasma. The plot on the right demonstrates how the mass defect plot can be used to visualize data and mine using Kendrick formulas, for example unknown polymer identification. All data is interactively linked between plots and data tables within Compound Discoverer software to streamline data review.
Compound Discovererソフトウェアを使用して、食品の不純物および分解生成物の代謝運命や構造を解析し、土壌や水に含まれる環境汚染物質を検出できます。環境および食品の安全性に関する研究において未知化合物が同定されると、多くの場合は四重極または高分解能 MS 法を用いたハイスループットスクリーニングが必要になります。Compound Discovererソフトウェアでは、新規または既存のmzVaultライブラリもしくはThermo Scientific TraceFinderソフトウェアで使用する標的リストにデータを直接エクスポートしてスクリーニングや定量を実施できるため、組織におけるメソッド移行の負担を減らせます。
Compound Discovererソフトウェアで乱用薬物や構造的に関連するデザイナードラッグの未知代謝物を検出できます;たとえば、多くの新規薬物が類似構造を有していますが、化合物クラススコアリングノードを使用すれば、検出された化合物の共通するフラグメントイオンに対してスコアを付けることで、特徴的なフラグメントから新たな薬物を見つけるのに役立ちます。この情報はスクリーニングメソッドに移行できるため、増加し続ける新規薬物とその代謝物に対応することができます。Compound Discovererソフトウェアが有する複数のワークフローのいずれかを用いて未知化合物を同定できたら、そのデータを新規または既存のmzVaultライブラリ、もしくはThermo Scientific TraceFinderソフトウェアで使用可能な標的リストにエクスポートし、四重極または高分解能MS法を用いてスクリーニングや定量を行えます。
* 法医学用途専用
Compound Discovererソフトウェア活用ツール
包括的なデータセットを理解して解釈する際に役立つツールがいくつかあります。Compound Discovererソフトウェアは多数のオンラインおよびオフラインのリソースにアクセスできるうえ、完全に一致するスペクトルが見つからない場合には、インテリジェントアルゴリズムを使用して未知化合物の同定をサポートします。
- mzCloudは広範かつ高度なオンラインマススペクトルフラグメンテーションデータベースです。
- mzLogicは、使用可能な数百万の構造データベースとmzCloudの広範な質量分析フラグメンテーションライブラリを組み合わせたデータ解析アルゴリズムであり、完全に一致するスペクトルがない場合に、未知化合物の推定構造をランク付けします。
- mzVaultは、オンラインにアクセスできない場合や自分の専有ライブラリを使用する必要がある場合に使えるリポジトリです。mzCloud のMS/MSレベルの内容にアクセスでき、カスタムローカルライブラリを作成できます。
- 多変量または単変量の統計解析のための統計解析およびデータ標準化ツールもあります。
同定した化合物をこれらのツールを用いて紐づけることができるため、解析の次の段階で使用するためにデータを選択してさまざまなソースにエクスポートするのが容易になります。

Click to enlarge
You can direct creation or expansion of mzVault spectral libraries or TraceFinder software format lists using text lists, mass lists, inclusion and exclusion lists, enabling you to streamline your analyses and subsequent targeted screening and/or quantitative analysis with minimal effort.
You can direct creation or expansion of mzVault spectral libraries or TraceFinder software format lists using text lists, mass lists, inclusion and exclusion lists, enabling you to streamline your analyses and subsequent targeted screening and/or quantitative analysis with minimal effort.
Compound Discovererで使用できる強力な活用ツールの詳細

mzCloud は低分子アプリケーションに幅広く対応し、多様な構造および化合物情報をそろえているため、非常に高い信頼性で未知化合物の同定を行えます。
網羅的な高分解能MS/MSおよび多段階MSnスペクトルを、幅広いメタデータ、世界最大級のLC-MSnリファレンススペクトルライブラリ、そして非常に広範囲にキュレーションされたマススペクトルライブラリとあわせて活用し、未知化合物をパワフルに同定します。

オンラインデータベースまたはユーザーが提供した構造から構造検索できる機能を存分に活用して、MSnおよび部分構造のスペクトルマッチからより多くの未知化合物を高い信頼性で同定できます。
世界最大級のマススペクトルフラグメンテーションライブラリであるmzCloudはどのようにして作成されたのですか?
多数のプリカーサーイオンおよびMSnフラグメンテーションスペクトルをmzCloud内の各化合物のスペクトルツリーに論理的に整理しています。スペクトルツリーの各レベルはMSnの段階を表しており、トップレベルはn=1、つまりプリカーサーイオンのスペクトルから始まります。その後生じるフラグメントの代表的なスペクトルを幅広くカバーして高品質な検索結果が得られる可能性を高めるために、さまざまな実験条件下でデータを取得するので、各レベルに多数のスペクトルを収容できるようになっています。

これは mzCloud のスペクトルツリーの模式図です。量極性の化合物(ESI +/-)や、さまざまなアダクトのMSスペクトルを取得しています。異なるフラグメンテーション法(CID、HCD)を用い、複数の衝突エネルギーで各プリカーサーイオンを完全にフラグメント化し、フラグメンテーションレベルごとにフラグメンテーションスペクトルのコレクションを作成します(MS2、MS3、MS4など)。その結果、各ライブラリエントリーについて網羅的なスペクトルツリーが生成されます。
化合物を正確に同定するうえで、各ライブラリエントリーについての詳細なデータは非常に重要なため、実測データをライブラリの内容とマッチさせ、Compound DiscovererおよびMass Frontierデータ解析ソフトウェアパッケージにより一致の信頼度を取得してデータを視覚化します。その他のツールとしてはmzLogicがあり、スペクトルライブラリの化合物エントリーのみでは同定できなかった未知化合物を、詳細なフラグメンテーション情報を使用して高い信頼性で同定します。

ライブラリ検索で一致するものが見つからなかった場合は?そんなときも、mzCloudに収容されている網羅的なフラグメンテーション情報を活用できます!スペクトルの類似性と部分構造情報(プリカーサーイオンの特徴的なスペクトル)から、mzLogicがあらゆる情報を集め、まったくの未知化合物の最有力候補を提示します。
未知低分子のスペクトルがライブラリでヒットしない場合でも、それを同定するには?
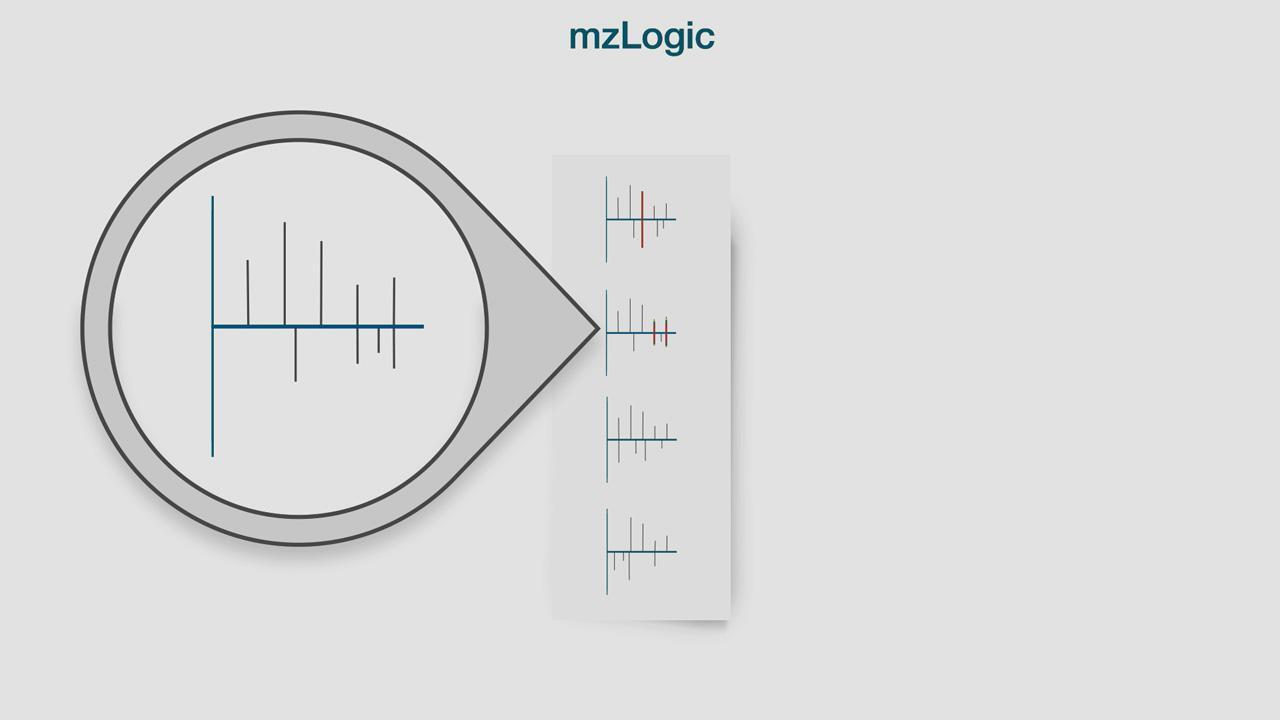
スペクトルライブラリの類似性検索を化学データベース検索と組み合わせることにより、実際のフラグメンテーションデータを最大活用。
Metabolikaで反応経路を作成、編集、検索できます。反応経路を作成および編集するための出版物レベルのグラフィックと、幅広い生物の370を超えるキュレーションおよびアノテーション済み生化学的経路により、経路に関する情報を簡単に共有できます。
Metabolikaの情報はフラグメンテーションの推測やmzLogicにも使用され、未知化合物同定の可能性をさらに高めます。

Click to enlarge
An example of biological pathway, which can be edited or added to, included in Metabolika.
An example of biological pathway, which can be edited or added to, included in Metabolika.
また、安定同位体標識分析では、置換率(組み込み率)をMetabolikaに入力し、対象経路をより包括的に見渡せます。

Click to enlarge
Combining stable isotope labelling with the visualization capabilities of Metabolika, allows you to overlay exchange rate information to provide a highly visual way of reviewing and reporting qualitative flux analyses.
Combining stable isotope labelling with the visualization capabilities of Metabolika, allows you to overlay exchange rate information to provide a highly visual way of reviewing and reporting qualitative flux analyses.
Metabolikaに加え、Compound DiscovererソフトウェアはKEGGおよびBioCyc生物学的経路データベースの両方に対応しています。化合物マッピングは次の2種類の方法で表示できます:コンテクスト特異的、すなわち特定の化合物を見る方法では、どの経路に当該化合物がマッピングされたかを確認できます。また、グローバルビューは、経路のリストから始める方法で、対象の経路にマッピングされたすべての化合物を表示できます。検出された化合物はmzCloudを用いて確認できます。たとえば色分けしたデータを登録された経路に表示します。
データとは、あなたが取得した知識であり、それだけで価値のあるものです。mzVaultではオフラインでmzCloud のMS2レベルのスペクトルデータにアクセスして検索したり、自前のスペクトルライブラリ情報を保管したりできます。スペクトル情報とあなたが得た知識は自動的にCompound Discovererから新規または既存のライブラリに送信され、Compound Discoverer またはTraceFinderソフトウェアを用いて検索したり、Thermo Scientific Mass Frontierソフトウェアを用いて編集したりできます。
広範な構造データベースや、構造または部分構造を提示するmzLogicを用いても、未知化合物を同定できない場合があります。こうした情報を過去に同定した化合物のライブラリとともに保管しておくと、「この化合物を扱ったことがあるか?」という疑問に答えることができて便利です。
Compound Discovererソフトウェアは、新規の環境汚染物質からデザイナードラッグおよび代謝物まで多くのアプリケーションにおいて、未知化合物を高い信頼性で同定する手段を提供します。こうしたアプリケーションでは、次のステップとしてTraceFinderソフトウェアと四重極または高分解能MSを用いた、より高いスループットでの同定および/または定量、もしくは第三者機関のパッケージによるさらに詳しい分析を行う場合があります。
適切な実験デザインはどのような分析においても非常に重要ですが、認められた傾向が実験の影響によるものではなく、実際の変化によるものであることを保証しなければならない統計学的研究においては特に重要です。したがって、大規模研究のプロトコルではpooled QC(品質管理)サンプルを活用して大規模研究の標準化を行うことがあります。

Click to enlarge
Using pooled QC samples, which are analyzed throughout data acquisition, allows for the correction of batch-effects over time. Correction for each sample is performed individually, the upper plot shows a curve fitted to the QC samples, with the bottom plot showing the resulting data set after correction. This capability is based upon a peer-reviewed methodology published by Dunn et. al. in Nature Protocols. Compound Discoverer software provides the capability to also view the impact of any changes to the data pre- and post- normalization according to this protocol.
Using pooled QC samples, which are analyzed throughout data acquisition, allows for the correction of batch-effects over time. Correction for each sample is performed individually, the upper plot shows a curve fitted to the QC samples, with the bottom plot showing the resulting data set after correction. This capability is based upon a peer-reviewed methodology published by Dunn et. al. in Nature Protocols. Compound Discoverer software provides the capability to also view the impact of any changes to the data pre- and post- normalization according to this protocol.
Compound Discovererソフトウェア内の強力で幅広い統計ツールはすべて互いにリンクしており、データ中のどの化合物/グループがどれくらい変化したかを理解するのに役立ちます。
統計解析は、メタボロミクス、環境、食品の安全性および異物混入、法医学、臨床、不純物、抽出物および浸出物(Extractables & Leachables)の研究など、さまざまな分析に幅広く使用できます。差分分析からANOVA、PCA、PLS-DA まで、さまざまな単変量および多変量解析を行うことができ、これらのツールで得られたアウトプットと、ワークフローで得られた化合物同定の結果とを、わかりやすいグラフィックとインタラクティブな方法により組み合わせることでデータから詳細な知見が得られ、それを簡単に報告、共有できます。

Click to enlarge
Demonstrating the connectivity of data within Compound Discoverer software, the data points highlighted by blue circles in the Volcano Plot (bottom right) are selected within the Compound Table (bottom left). Selecting any compound in any plot automatically updates all plots to show the relevant data. The interconnected tools enable you to rapidly identify differences and the compounds or groups of compounds responsible for those differences. Additionally, it streamlines follow-on confirmation by giving you the ability to filter and review the relevant data.
Demonstrating the connectivity of data within Compound Discoverer software, the data points highlighted by blue circles in the Volcano Plot (bottom right) are selected within the Compound Table (bottom left). Selecting any compound in any plot automatically updates all plots to show the relevant data. The interconnected tools enable you to rapidly identify differences and the compounds or groups of compounds responsible for those differences. Additionally, it streamlines follow-on confirmation by giving you the ability to filter and review the relevant data.
Compound Discovererソフトウェアにより、複合的なデータセットとその関係性を視覚化できるため、モニター間で複数のプロットを追加し、こうした関係を追跡、表示してデータをより深く理解することができます。

Click to enlarge
From Principal Components Analysis (PCA) for unbiased review of data to supervised techniques like Partial Least Squares – Discriminant Analysis (PLS-DA) and the use of S Plots (left) identify compounds that give rise to any observed grouping of samples. Hierarchical Clustering (center) not only shows the clustering of samples along the x-axis, and clustering of compounds on the y-axis, it provides user-configurable heat mapping to visualize any clustering. Box Whisker Charts (right) allow visualization by groupings, time points, and more, with dynamic.
From Principal Components Analysis (PCA) for unbiased review of data to supervised techniques like Partial Least Squares – Discriminant Analysis (PLS-DA) and the use of S Plots (left) identify compounds that give rise to any observed grouping of samples. Hierarchical Clustering (center) not only shows the clustering of samples along the x-axis, and clustering of compounds on the y-axis, it provides user-configurable heat mapping to visualize any clustering. Box Whisker Charts (right) allow visualization by groupings, time points, and more, with dynamic.
複合的なデータセットを徹底的に確認し、差異を生じさせる構成因子を評価した後、その変化/差異が同定された化合物によるものであるかを検証するために、より大規模な解析が必要になることもあります。確認済みの化合物はCompound Discovererソフトウェアからさまざまな形で簡単にエクスポートできるため、追加の分析を実施しやすくなります。詳細は、「カスタムローカルライブラリおよびデータ転送」セクションを参照してください。
化合物どうしの関係性を調べることにより、データセットに関してさらなる情報や知見が得られます。分子ネットワークを用いれば、構造変化やスペクトルの類似性に基づく解析において、たとえば、Phase IおよびPhase IIのさまざまな生体内変換など、化合物間の関係をインタラクティブに探索することができます。

Click to enlarge
The fully interactive Molecular Networks visualization browser allows you to view your data in a different way. Identified compounds are shown by nodes (circles) and when a relationship is identified, the nodes are connected. Selecting a node (compound) or connection (transformation) displays pertinent information (right) about the identified compound and the relevant transformation(s). All of the visualized data can be interactively filtered using thresholds, data quality information or text search for specific compounds or transformations.
The fully interactive Molecular Networks visualization browser allows you to view your data in a different way. Identified compounds are shown by nodes (circles) and when a relationship is identified, the nodes are connected. Selecting a node (compound) or connection (transformation) displays pertinent information (right) about the identified compound and the relevant transformation(s). All of the visualized data can be interactively filtered using thresholds, data quality information or text search for specific compounds or transformations.
FISh(フラグメントイオンサーチ)は、理論的なフラグメントの推測または実測MSnデータから得られた親化合物のフラグメンテーションパターンに基づき、構造が類似している化合物を迅速にスクリーニングする方法です。親化合物の構造およびそこから生じうる代謝物を用いて、マトリックス関連バックグラウンドイオンの大部分をフィルタリングして取り除き、関係のある化合物を迅速かつ容易に同定します。FIShによりPhase lおよびPhase llの生体内変換の詳細なリストが得られ、リストのカスタマイズもできます。

Click to enlarge
In addition to filtering the structurally similar compounds, FISh automatically localizes transformation sites, labels, and applies color-coding to fragments common to the parent and filtered results. In the Mirror Plot example shown here, exact matches to proposed metabolite fragments are shown in green and transformation shifted matches are blue.
In addition to filtering the structurally similar compounds, FISh automatically localizes transformation sites, labels, and applies color-coding to fragments common to the parent and filtered results. In the Mirror Plot example shown here, exact matches to proposed metabolite fragments are shown in green and transformation shifted matches are blue.
HighChem フラグメンテーションライブラリは、査読付き論文に掲載されている 52,000を超えるフラグメンテーションスキーム、217,000の個別の反応、256,000の化学構造および216,000の解読済みメカニズムから得られる情報が登録されていますが、FIShはこのライブラリを採用しているため、推定代謝物への構造の割当てやその他の構造の予測に役立つ強力なツールです。FISh では、推定構造のフラグメンテーション構造を提示する際の信頼性を向上させるために、実際のデータを使用し、提示された候補構造によってフラグメンテーションデータをどれだけ説明できるかを表すスコアを算出します。

このホワイトペーパーでは、マススペクトルライブラリによって低分子同定の課題を解決します。mzCloudスペクトルライブラリ、およびmzVaultソフトウェアは、ルーチンおよび研究アプリケーションにおける低分子同定の課題に対応できるようデザインされています。
Compound Discovererソフトウェア動画
Compound Discovererソフトウェアのパワフルな特長について、ユーザーや科学者から詳細な説明を聞くには、以下の動画をご覧ください。

低分子特性評価や同定が意思決定を鈍らせていますか?質量分析ソフトウェアなどのクラウドベース技術はますますラボラトリーに普及しています。明日の問題を今日解決しましょう。
Compound Discovererソフトウェア注文ガイド
関連ソフトウェアアプリケーションおよびテクノロジー

質量分析装置およびソフトウェアに関するすべてのサポートニーズに対応するリソースです。実験開始時や、一般的な問題への答えをお探しの際に、関連する技術情報を取得したり、ヒントやコツをご覧になれます。