Search Thermo Fisher Scientific
反转录——六种最常见的应用

在哪里会使用到反转录?
反转录通常用于研究 RNA 的功能。RNA 通常通过反反转录 (RT) 转化为更稳定的互补 DNA (cDNA)。获得的 cDNA 可用于使用基于 DNA 的技术 (如克隆、, PCR 和测序) 进一步研究 RNA ,因此反反转录是许多基于 RNA 的实验研究工作流程中的关键步骤。

为了研究RNA的功能,通常通过逆转录(RT)将RNA转化为更稳定的互补DNA(cDNA)。cDNA可通过克隆、PCR和测序等技术研究RNA,因此,逆转录是许多RNA实验工作流程的关键步骤。
反转录聚合酶链式反应 (RT-PCR)
在 RT-PCR 中,通过反转录 (RT) 将 RNA 转化为 cDNA,然后 cDNA 通过聚合酶链式反应 (PCR)进行扩增(图1)。利用 cDNA 扩增步骤,有望对初始 RNA 进行进一步研究,即便 RNA 样本数量有限或低丰度表达。RT-PCR 的常见应用包括检测表达基因、,转录本变异检测变体检查,以及生成用于克隆和测序所用的 cDNA 模板的制备。
")
图1.反转录聚合酶链式反应 (RT-PCR)。RT = 反转录, RTase = 反转录酶。
由于反转录为 PCR 扩增和下游实验提供 cDNA 模板,因此是实验成功的关键步骤之一。所选择的反转录酶应具有较高的反转录销量,即便针对具有挑战性的 RNA 样本,比如已降解样本、含抑制剂样本或具有高度二级结构的样本。
在进行 RT-PCR 时,一步法和两步法是常用的两种方法,每种方法都具有各自的优缺点。顾名思义,一步法 RT-PCR将第一链 cDNA 合成 (RT) 和后续 PCR 结合在单个反应管中。这种反应体系配置可简化实验流程,降低结果变异,并尽可能减少可能的污染。两步法 RT-PCR 包含两个独立反应,首先进行第一链 cDNA 合成 (RT) ,然后在另一个单独的反应管中扩增第一步所得的 cDNA。因此,两步法 RT-PCR 可用于检测单个 RNA 样品中的多个基因。
了解更多信息:一步法 RT-PCR 与两步法 RT-PCR
反转录—定量 RT-PCR 的第一步
RT-qPCR 最常见应用之一是测定 mRNA 水平随时间、空间或特殊处理后 (如药物处理) 的变化。由于 RT-qPCR 的灵敏度高于 RT-PCR , RT-qPCR 也广泛用于检测是否存在逆转录病毒 (RNA 病毒)。与 RT-PCR 实验流程类似, RNA 首先转化为 cDNA ,然后通过 PCR 扩增。主要的区别在于,在指数扩增阶段,RT-qPCR 是通过荧光实时检测 cDNA 扩增的水平。此扩增水平作为对 RNA 中初始靶标进行定量的基准。
了解更多信息:定量 PCR
博文:了解实时荧光定量 PCR 中的 Ct 值
视频:反转录酶对 Ct 值的影响
了解 qPCR 中 Ct 值的含义,哪些因素会影响 Ct 值以及如何选择反转录酶以改善 Ct 值。
视频:克服 RT-qPCR 中的挑战
了解使用 RT-qPCR 进行基因表达分析中出现变异的五大原因,以及克服常见 RT 失误和挑战的技巧。
通过 RT-qPCR 进行基因表达定量检测的准确性在很大程度上取决于 cDNA 模板的质量和数量。因此,反转录步骤对于 RT-qPCR 的成功至关重要。反转录步骤应产生能代表初始 RNA 的 cDNA 产物。因此,所选择的反转录酶应该具有高效合成 cDNA 的能力,即使对于低丰度基因和纯度不佳或复杂 RNA 样本 (如高 GC 含量、含抑制剂、降解样本) 。
了解更多信息:反转录酶特性
应用说明: 针对植物样本改进的 RT-qPCR 分析
应用说明: 针对全血 RNA 样本改进的 RT-qPCR 分析
除了高效的反转录酶外,选择用于 RT 反应的试剂时还有许多考虑因素。首先,在较宽的起始量 RNA 范围内,cDNA 的动态范围或线性扩增至关重要。获取与起始 RNA 量成比例 cDNA 产量的能力可确保对基因表达的准确定量分析(图2)。
白皮书: 改进型反转录酶

图2.在一系列不同起始量总 RNA,使用 RT 预混液扩增后 qPCR 结果的线性度,用于检测 (A) 高丰度和 (B) 低丰度 RNA 靶标。RNA 起始量从 10 μg 到 1 μg ,经反转录,随后通过 PCR 扩增。两种预混液均生成与 RNA 上样 量成比例的 cDNA,但如较低 (即早期) CT 值所示,从预混液 1 中获得更高的产量,尤其是对于低丰度基因靶标。
此外,所选的试剂应能在重复实验中获得大量且一致的 cDNA 得率,以获得灵敏度高且差异性小的基因表达结果 (图 3)。含有反转录所需所有成分的单管预混液有助于尽可能减少实验变量、交叉污染和移液失误。
白皮书: 经优化的 RT-qPCR 预混液

图3.使用不同 RT 预混液后 qPCR 结果的灵敏度和变异性,用于检测 (A) 高丰度和 (B) 低丰度 RNA 靶标。在这些试剂中, 30 次重复实验后,预混液 1 的平均 值和标准偏差最低,说明反转录试剂的选择对进行可靠基因表达分析的重要性。
一种特殊的 RT-qPCR 是直接从粗细胞裂解物中进行反转录,而无需分离 RNA [1]。在研究少量细胞的实验中,使用稀缺的样或选择特定细胞时,可考虑进行直接 RT-qPCR 以防止潜在样本损失和低 RNA 回收率。在直接 RT-qPCR 中,抑制内源性 RNase(其可降解 RNA)和在细胞裂解过程中去除细胞基因组 DNA 非常重要。使用针对低丰度靶标而优化的试剂盒,样品制备可以在短短 7 分钟内完成,同时可从。 具有高持续合成能力的反 转录酶尤其适用于对未纯化 RNA 提取物的反转录,因为其具有对抑制剂较高的耐受性和高灵敏度。
cDNA 克隆和文库构建中反转录的应用
分子生物学中反转录酶最高的应用之一就是构建 cDNA 文库 [2 – 4]。cDNA 文库由代表特定样本中转录序列的 cDNA 克隆组成。因此,文库可提供针对特定细胞类型、器官或发育阶段基因的时间和空间表达信息。cDNA 文库克隆用于新型 RNA 转录本的表征、基因序列测定和重组蛋白的表达。
构建 cDNA 文库的关键在于是否能对 RNA 全长和相对丰度的合理表征,因此反转录酶的选择非常重要。具有高持续合成能力的反转录酶可合成全长 cDNA 并捕获低丰度 RNA。同样,具有高热稳定性的反转录酶推荐用于含复杂二级结构 RNA 的逆转录。
反转录后,可使用多种方法将 cDNA 插入 载体 中以进行克隆。第二链 cDNA 合成 后的双链 cDNA 通常是平末端,可直接克隆进入平末端载体 (图 4A)。尽管这种方法涉及的步骤更少,但平末端克隆可能连接效率较低且插入后失去方向性。
了解更多信息:克隆实验流程
或者,可以对 cDNA 末端进行修饰,以包括已知序列的其他核苷酸。例如,为修饰 cDNA 的 5′ 末端,可以使用具有额外 5′ 核苷酸的oligo(dT) 引物来启动反转录;为修饰 cDNA 的 3′ 末端,可以连接具有所需序列的短 DNA 寡核苷酸,又称为 linker 或 adapter (图 4B)。通过这种方式,定向插入(例如限制性和同源重组)、启动子结合(例如 T3 和 T7 序列)和亲和纯化(例如生物素和 His 标签)的位点可以很容易地掺入 cDNA 序列中。

图4.克隆 cDNA 的常用方法。(A) 具有平末端的双链 cDNA 可直接克隆到平末端克隆载体中。(B) 对于定向克隆, cDNA 末端可使用与载体兼容的独特序列进行修饰。(C) 可使用互补的末端序列进行不依赖连接酶的克隆,以提高插入效率。(D) 当插入片段的序列已知时,可考虑通过 PCR 进行基因特异性克隆。
在另一种常用策略中,可以使用互补同聚物加尾对 cDNA 插入片段和载体的 3′ 末端进行酶促延伸。使用 末端脱氧核苷酸转移酶 (TdT) 和单一 dNTP,可以在插入片段上添加一串含 20 – 30 个核苷酸的序列,并将一串相似的互补核苷酸添加到载体上 (如插入片段上的 Cs 和载体上的 GS) ,使载体和插入片段尾部相互退火 (图 4C)。在细菌,因此无需连接步骤。
若目标序列是已知的,可通过 RT-PCR 生成插入片段,用于克隆 cDNA的特定区域 (图 4D)。
了解更多信息:PCR 克隆
cDNA 末端快速扩增 (RACE) 中的反转录应用
cDNA 末端快速扩增 (RACE) 是一种基于 PCR 的方法,可用于确定 cDNA 5′ 和 3′ 末端的未知序列 [5]。这些方法通常被称为 5′ RACE 和 3′ RACE。RACE 的实验目的包括鉴定 5′ 和 3′ 非翻译区,研究异质性转录起始位点,表征启动子区域,测定完整 cDNA 序列 ,以及对用于蛋白表达的完整开放阅读框 (ORF) 进行测序。
使用具有单侧特异性的 PCR (也称为单侧或锚定 PCR [6 , 7]) 扩增 cDNA 的未知区域作为 RACE 产物。5′ RACE 依赖于带寡核苷酸的 5′ 末端延伸,用于 PCR 引物结合,而 3′ RACE 利用 mRNA 的 poly(A) 尾作为 PCR 的通用引物位点 (图 5)。

点击放大图片
图 5.5′ 和 3′ RACE。
在 5′ RACE (图 5A) 中,使用基因特异性引物,将特定序列或相关家族的 mRNA 反转录为第一链 cDNA。然后使用 末端脱氧核苷酸转移酶 (TdT)通在 cDNA 的 3′ 末端添加一个同聚物尾部结构 (通常是一串 Cs),或将 cDNA 3′ 末端连接到寡核苷酸接头上。随后,进行两轮半嵌套 PCR,以扩增具有 5′ 未知序列的区域。还可使用下游应用的引物通过 PCR 对扩增子进行末端延伸,例如用于定向克隆的限制性位点引入和用于测序的通用测序引物结合位点。
在 3′ RACE (图 5B) 中,使用含接头序列的 oligo (dT) 引物将 mRNA 反转录为 cDNA。然后,使用已知上游外显子序列的特异性引物和通过 oligo (dT) 引物引入的接头序列,进行两轮半嵌套 PCR。通过这种方式,可对外显子和 poly(A) 尾之间的未知 3´ mRNA 序列进行扩增,以供进一步分析。
起始 RNA 的质量和反转录反应体系配制对于成功进行 RACE 实验至关重要。在 5′ RACE 中,任何长度 (甚至包括未达 mRNA 5′ 末端的产物) 的第一链 cDNA 都将具有添加序列 (即同聚物尾部结构或接头) ,随后通过 PCR 进行扩增。为了最大限度合成全长 cDNA ,应选择具有最小 RNase H 活性、高持续合成能力和高热稳定性的反转录酶。
了解更多信息:反转录酶特性
基因表达芯片中的反转录应用
1990 年代期间,DNA 芯片的发展开启了大规模无偏差或先前假说的基因表达图谱分析。芯片由玻璃或硅晶片上数千个被称为“features”或“spots”的腔室组成。每个腔室的表面上固定有相同拷贝数的单链DNA序列,称为“探针”,每个探针代表一个基因。探针与用于芯片的荧光标记 cDNA 靶标杂交,可同时比较两个样品之间的基因表达 (图 6 和 7) [10-12]。

图 6.基因表达微阵列芯片。
芯片探针由生物体已知基因组序列或 cDNA 生成。例如,可以利用 PCR 对每个已知基因进行扩增,然后将其产物变性为单链 DNA ,并将其固定到芯片上作为探针。或者,可直接在芯片上合成 20 – 60 nt 的寡核苷酸作为芯片探针 [13]。
图 7 概述了微阵列芯片如何用于不同基因的表达分析。首先,从两个实验样本(也称为“检测样本”或“处理样本)和对照样本(也称“参考样本”或“正常样本”)中分离总 RNA 或 mRNA。然后将纯化的 RNA 样本转化为 cDNA ,并使用不同的荧光染料进行标记。接下来,将两个样本的标记 cDNA 靶标混合,并与一个微阵列芯片上的探针杂交。洗去未结合的靶标后,扫描芯片以检测标记的荧光基团。然后分析两种荧光信号的比例,以定量测定受实验条件影响的基因的表达。
立即查看:微阵列分析

点击图片放大
图7.基因表达芯片的 cDNA 靶标制备和分析。
cDNA 靶标可以在反转录过程中或反转录后进行标记 (图 8)。在直接标记中,在 cDNA 合成过程中即掺入荧光标记的核苷酸。在间接标记中,先使用已修饰的核苷酸进行反转录,然后用荧光基团标记 cDNA。尽管间接标记方法需要更长的实验流程,但荧光标记效率往往更高。

图8.直接和间接 cDNA 标记。
当 RNA 起始量较少 (如 10 – 100 ng) 时,可使用 T7- oligo (dT) 启动子引物将 RNA 反转录为双链 cDNA。然后通过体外转录 (图 9)。在体外转录过程中,可以使用修饰核苷酸直接或间接标记 RNA。或者,对扩增的 RNA 进行反转录并标记以生成 cDNA 靶标。

点击图片放大
图9.通过将 RNA 转化为 cDNA,然后从添加的启动子序列进行体外转录,实现 RNA 的扩增。
用于 RNA 测序 (RNA-Seq) 的反转录应用
RNA 测序,又称为 RNA-Seq, 通常可用于研究从基因组转录的 RNA 及其调控机制。随着二代测序 (NGS) 的出现, RNA-Seq 已成为分析全转录组 (即已转录的编码和长链非编码 RNA ) 、检测基因表达、发现剪接变异和融合转录本以及检测低丰度基因的高通量分析方法 [14,15] 。与基因芯片相比,RNA-Seq 的优点包括动态范围更大,灵敏度更高以及可在无基因组信息的情况下表征 RNA 序列。
由于大多数测序平台是为 DNA 设计的,因此 RNA-Seq 的模板制备需要进行反转录。最理想的结果是,所得到的 cDNA 能够无偏差代表初始 RNA,包括低丰度转录物。全长 cDNA 合成对于捕获样本中的所有 RNA 序列也非常重要。反转录的错误率也至关重要,这取决于测序文库大小和数据质量。因此,应谨慎认真选择反转录酶。
了解更多信息:反转录酶特性
研究目标和测序技术将决定 RNA-Seq 模板制备的顺序和方法 [16, 17]。尽管如此,典型的测序文库建库流程包括目标 RNA 的富集、RNA 或 cDNA 的片段化、 反转录 、测序接头 (在多重测序中,还需添加索引或条形码) 以及可选的 文库 PCR 扩增 (图 10)。

图10.RNA 测序的传统实验流程。
了解更多信息:RNA 测序
反转录环介导等温扩增 (RT-LAMP)
RT-LAMP 是一种快速、简单、灵敏的 RNA 检测方案,可使用多种方法用来评估检测结果。由于其具有简单的工作流程和快速的反应时间,尤其适用于在实验室或现场环境中对病毒病原体的检测和监测。
LAMP 方法依赖于具有强链置换活性的 DNA 聚合酶、经特殊设计的内外引物以及环状引物。对于 RNA 靶标的扩增,只需将反转录酶和 RNase 抑制剂添加到 LAMP 反应 (RT-LAMP) 中即可进行一步反应。
LAMP 在恒定温度 (60 – 65°C) 下进行,被归类为等温扩增。LAMP 扩增技术需要 4 或 6 个特殊设计的引物,该引物可与两个不同的靶标区域结合 (相隔~300 bp)。LAMP 最初被开发时使用 4 个引物,随后添加两个环状引物使反应时间缩短了一半。目前,使用 LAMP 法可在 15 分钟内扩增靶标 RNA 或 DNA。LAMP 所需的引物包括两个外部 (F3 和 B3) 引物、两个内部引物 (正向内引物 (FIP) 和反向内引物 (BIP) 和环状引物(正向环状引物 (Loop F) 和反向环状引物 (Loop B))。
LAMP 反应分两步进行:非循环步骤和自动循环步骤。第一步是内引物 (FIP) 与靶标 DNA 结合,开始互补链合成。之后紧接着是外引物 (F3) 的链侵入延伸,释放单链 DNA 作为下游引物的模板。转换得到的内部序列在 F 端形成一个茎环结构。在另一端借助 BIP 引物和 B3 引物进行同样的过程,最终获得在 3′ 和 5′ 端带有茎环的哑铃结构该结构包含多个用于重复扩增的起始位点,并通过自动循环加快了 DNA 扩增,从而得到不同长度和菜花样结构的扩增 DNA (图 11)。

图11.等温 DNA 扩增 (简化示意图) —引物 (3 对) :FIP/BIP、F3/BIP 和 Loop F/B。 靶标序列对茎环的形成比较重要;“ c ”代表互补 (例如,F1c 与 F1 互补)。
RT-LAMP 的优点
- 不需要热循环仪 (仅需加热模块)
- 快速周转时间 (15-60 分钟)
- 多种方法解读检测结果
- 简化的工作流程
- 对抑制剂具有高耐受性
- 适合用于现场检测
RT-LAMP 仅需要少量 RNA,耐受反应抑制剂,易于操作,特异性和灵敏度高。等温条件下的扩增无需热循环仪,扩增效率更高,无需等待温度变化。由于这些特性, LAMP 技术自其发现以来其应用经历了指数级增长。RT-LAMP 在实验室中用于快速检测病原体 (细菌、寄生虫和病毒)。由于其简单性,它也是适应现场或床旁检测的重要方法。
立即查看:LAMP 解决方案
反转录聚合酶链式反应 (RT-PCR)
在 RT-PCR 中,通过反转录 (RT) 将 RNA 转化为 cDNA,然后 cDNA 通过聚合酶链式反应 (PCR)进行扩增(图1)。利用 cDNA 扩增步骤,有望对初始 RNA 进行进一步研究,即便 RNA 样本数量有限或低丰度表达。RT-PCR 的常见应用包括检测表达基因、,转录本变异检测变体检查,以及生成用于克隆和测序所用的 cDNA 模板的制备。
图1.反转录聚合酶链式反应 (RT-PCR)。RT = 反转录, RTase = 反转录酶。
由于反转录为 PCR 扩增和下游实验提供 cDNA 模板,因此是实验成功的关键步骤之一。所选择的反转录酶应具有较高的反转录销量,即便针对具有挑战性的 RNA 样本,比如已降解样本、含抑制剂样本或具有高度二级结构的样本。
在进行 RT-PCR 时,一步法和两步法是常用的两种方法,每种方法都具有各自的优缺点。顾名思义,一步法 RT-PCR将第一链 cDNA 合成 (RT) 和后续 PCR 结合在单个反应管中。这种反应体系配置可简化实验流程,降低结果变异,并尽可能减少可能的污染。两步法 RT-PCR 包含两个独立反应,首先进行第一链 cDNA 合成 (RT) ,然后在另一个单独的反应管中扩增第一步所得的 cDNA。因此,两步法 RT-PCR 可用于检测单个 RNA 样品中的多个基因。
了解更多信息:一步法 RT-PCR 与两步法 RT-PCR
反转录—定量 RT-PCR 的第一步
RT-qPCR 最常见应用之一是测定 mRNA 水平随时间、空间或特殊处理后 (如药物处理) 的变化。由于 RT-qPCR 的灵敏度高于 RT-PCR , RT-qPCR 也广泛用于检测是否存在逆转录病毒 (RNA 病毒)。与 RT-PCR 实验流程类似, RNA 首先转化为 cDNA ,然后通过 PCR 扩增。主要的区别在于,在指数扩增阶段,RT-qPCR 是通过荧光实时检测 cDNA 扩增的水平。此扩增水平作为对 RNA 中初始靶标进行定量的基准。
了解更多信息:定量 PCR
博文:了解实时荧光定量 PCR 中的 Ct 值
视频:反转录酶对 Ct 值的影响
了解 qPCR 中 Ct 值的含义,哪些因素会影响 Ct 值以及如何选择反转录酶以改善 Ct 值。
视频:克服 RT-qPCR 中的挑战
了解使用 RT-qPCR 进行基因表达分析中出现变异的五大原因,以及克服常见 RT 失误和挑战的技巧。
通过 RT-qPCR 进行基因表达定量检测的准确性在很大程度上取决于 cDNA 模板的质量和数量。因此,反转录步骤对于 RT-qPCR 的成功至关重要。反转录步骤应产生能代表初始 RNA 的 cDNA 产物。因此,所选择的反转录酶应该具有高效合成 cDNA 的能力,即使对于低丰度基因和纯度不佳或复杂 RNA 样本 (如高 GC 含量、含抑制剂、降解样本) 。
了解更多信息:反转录酶特性
应用说明: 针对植物样本改进的 RT-qPCR 分析
应用说明: 针对全血 RNA 样本改进的 RT-qPCR 分析
除了高效的反转录酶外,选择用于 RT 反应的试剂时还有许多考虑因素。首先,在较宽的起始量 RNA 范围内,cDNA 的动态范围或线性扩增至关重要。获取与起始 RNA 量成比例 cDNA 产量的能力可确保对基因表达的准确定量分析(图2)。
白皮书: 改进型反转录酶
图2.在一系列不同起始量总 RNA,使用 RT 预混液扩增后 qPCR 结果的线性度,用于检测 (A) 高丰度和 (B) 低丰度 RNA 靶标。RNA 起始量从 10 μg 到 1 μg ,经反转录,随后通过 PCR 扩增。两种预混液均生成与 RNA 上样 量成比例的 cDNA,但如较低 (即早期) CT 值所示,从预混液 1 中获得更高的产量,尤其是对于低丰度基因靶标。
此外,所选的试剂应能在重复实验中获得大量且一致的 cDNA 得率,以获得灵敏度高且差异性小的基因表达结果 (图 3)。含有反转录所需所有成分的单管预混液有助于尽可能减少实验变量、交叉污染和移液失误。
白皮书: 经优化的 RT-qPCR 预混液
图3.使用不同 RT 预混液后 qPCR 结果的灵敏度和变异性,用于检测 (A) 高丰度和 (B) 低丰度 RNA 靶标。在这些试剂中, 30 次重复实验后,预混液 1 的平均 值和标准偏差最低,说明反转录试剂的选择对进行可靠基因表达分析的重要性。
一种特殊的 RT-qPCR 是直接从粗细胞裂解物中进行反转录,而无需分离 RNA [1]。在研究少量细胞的实验中,使用稀缺的样或选择特定细胞时,可考虑进行直接 RT-qPCR 以防止潜在样本损失和低 RNA 回收率。在直接 RT-qPCR 中,抑制内源性 RNase(其可降解 RNA)和在细胞裂解过程中去除细胞基因组 DNA 非常重要。使用针对低丰度靶标而优化的试剂盒,样品制备可以在短短 7 分钟内完成,同时可从。 具有高持续合成能力的反 转录酶尤其适用于对未纯化 RNA 提取物的反转录,因为其具有对抑制剂较高的耐受性和高灵敏度。
cDNA 克隆和文库构建中反转录的应用
分子生物学中反转录酶最高的应用之一就是构建 cDNA 文库 [2 – 4]。cDNA 文库由代表特定样本中转录序列的 cDNA 克隆组成。因此,文库可提供针对特定细胞类型、器官或发育阶段基因的时间和空间表达信息。cDNA 文库克隆用于新型 RNA 转录本的表征、基因序列测定和重组蛋白的表达。
构建 cDNA 文库的关键在于是否能对 RNA 全长和相对丰度的合理表征,因此反转录酶的选择非常重要。具有高持续合成能力的反转录酶可合成全长 cDNA 并捕获低丰度 RNA。同样,具有高热稳定性的反转录酶推荐用于含复杂二级结构 RNA 的逆转录。
反转录后,可使用多种方法将 cDNA 插入 载体 中以进行克隆。第二链 cDNA 合成 后的双链 cDNA 通常是平末端,可直接克隆进入平末端载体 (图 4A)。尽管这种方法涉及的步骤更少,但平末端克隆可能连接效率较低且插入后失去方向性。
了解更多信息:克隆实验流程
或者,可以对 cDNA 末端进行修饰,以包括已知序列的其他核苷酸。例如,为修饰 cDNA 的 5′ 末端,可以使用具有额外 5′ 核苷酸的oligo(dT) 引物来启动反转录;为修饰 cDNA 的 3′ 末端,可以连接具有所需序列的短 DNA 寡核苷酸,又称为 linker 或 adapter (图 4B)。通过这种方式,定向插入(例如限制性和同源重组)、启动子结合(例如 T3 和 T7 序列)和亲和纯化(例如生物素和 His 标签)的位点可以很容易地掺入 cDNA 序列中。
图4.克隆 cDNA 的常用方法。(A) 具有平末端的双链 cDNA 可直接克隆到平末端克隆载体中。(B) 对于定向克隆, cDNA 末端可使用与载体兼容的独特序列进行修饰。(C) 可使用互补的末端序列进行不依赖连接酶的克隆,以提高插入效率。(D) 当插入片段的序列已知时,可考虑通过 PCR 进行基因特异性克隆。
在另一种常用策略中,可以使用互补同聚物加尾对 cDNA 插入片段和载体的 3′ 末端进行酶促延伸。使用 末端脱氧核苷酸转移酶 (TdT) 和单一 dNTP,可以在插入片段上添加一串含 20 – 30 个核苷酸的序列,并将一串相似的互补核苷酸添加到载体上 (如插入片段上的 Cs 和载体上的 GS) ,使载体和插入片段尾部相互退火 (图 4C)。在细菌,因此无需连接步骤。
若目标序列是已知的,可通过 RT-PCR 生成插入片段,用于克隆 cDNA的特定区域 (图 4D)。
了解更多信息:PCR 克隆
cDNA 末端快速扩增 (RACE) 中的反转录应用
cDNA 末端快速扩增 (RACE) 是一种基于 PCR 的方法,可用于确定 cDNA 5′ 和 3′ 末端的未知序列 [5]。这些方法通常被称为 5′ RACE 和 3′ RACE。RACE 的实验目的包括鉴定 5′ 和 3′ 非翻译区,研究异质性转录起始位点,表征启动子区域,测定完整 cDNA 序列 ,以及对用于蛋白表达的完整开放阅读框 (ORF) 进行测序。
使用具有单侧特异性的 PCR (也称为单侧或锚定 PCR [6 , 7]) 扩增 cDNA 的未知区域作为 RACE 产物。5′ RACE 依赖于带寡核苷酸的 5′ 末端延伸,用于 PCR 引物结合,而 3′ RACE 利用 mRNA 的 poly(A) 尾作为 PCR 的通用引物位点 (图 5)。
点击放大图片
图 5.5′ 和 3′ RACE。
在 5′ RACE (图 5A) 中,使用基因特异性引物,将特定序列或相关家族的 mRNA 反转录为第一链 cDNA。然后使用 末端脱氧核苷酸转移酶 (TdT)通在 cDNA 的 3′ 末端添加一个同聚物尾部结构 (通常是一串 Cs),或将 cDNA 3′ 末端连接到寡核苷酸接头上。随后,进行两轮半嵌套 PCR,以扩增具有 5′ 未知序列的区域。还可使用下游应用的引物通过 PCR 对扩增子进行末端延伸,例如用于定向克隆的限制性位点引入和用于测序的通用测序引物结合位点。
在 3′ RACE (图 5B) 中,使用含接头序列的 oligo (dT) 引物将 mRNA 反转录为 cDNA。然后,使用已知上游外显子序列的特异性引物和通过 oligo (dT) 引物引入的接头序列,进行两轮半嵌套 PCR。通过这种方式,可对外显子和 poly(A) 尾之间的未知 3´ mRNA 序列进行扩增,以供进一步分析。
起始 RNA 的质量和反转录反应体系配制对于成功进行 RACE 实验至关重要。在 5′ RACE 中,任何长度 (甚至包括未达 mRNA 5′ 末端的产物) 的第一链 cDNA 都将具有添加序列 (即同聚物尾部结构或接头) ,随后通过 PCR 进行扩增。为了最大限度合成全长 cDNA ,应选择具有最小 RNase H 活性、高持续合成能力和高热稳定性的反转录酶。
了解更多信息:反转录酶特性
基因表达芯片中的反转录应用
1990 年代期间,DNA 芯片的发展开启了大规模无偏差或先前假说的基因表达图谱分析。芯片由玻璃或硅晶片上数千个被称为“features”或“spots”的腔室组成。每个腔室的表面上固定有相同拷贝数的单链DNA序列,称为“探针”,每个探针代表一个基因。探针与用于芯片的荧光标记 cDNA 靶标杂交,可同时比较两个样品之间的基因表达 (图 6 和 7) [10-12]。
图 6.基因表达微阵列芯片。
芯片探针由生物体已知基因组序列或 cDNA 生成。例如,可以利用 PCR 对每个已知基因进行扩增,然后将其产物变性为单链 DNA ,并将其固定到芯片上作为探针。或者,可直接在芯片上合成 20 – 60 nt 的寡核苷酸作为芯片探针 [13]。
图 7 概述了微阵列芯片如何用于不同基因的表达分析。首先,从两个实验样本(也称为“检测样本”或“处理样本)和对照样本(也称“参考样本”或“正常样本”)中分离总 RNA 或 mRNA。然后将纯化的 RNA 样本转化为 cDNA ,并使用不同的荧光染料进行标记。接下来,将两个样本的标记 cDNA 靶标混合,并与一个微阵列芯片上的探针杂交。洗去未结合的靶标后,扫描芯片以检测标记的荧光基团。然后分析两种荧光信号的比例,以定量测定受实验条件影响的基因的表达。
立即查看:微阵列分析
点击图片放大
图7.基因表达芯片的 cDNA 靶标制备和分析。
cDNA 靶标可以在反转录过程中或反转录后进行标记 (图 8)。在直接标记中,在 cDNA 合成过程中即掺入荧光标记的核苷酸。在间接标记中,先使用已修饰的核苷酸进行反转录,然后用荧光基团标记 cDNA。尽管间接标记方法需要更长的实验流程,但荧光标记效率往往更高。
图8.直接和间接 cDNA 标记。
当 RNA 起始量较少 (如 10 – 100 ng) 时,可使用 T7- oligo (dT) 启动子引物将 RNA 反转录为双链 cDNA。然后通过体外转录 (图 9)。在体外转录过程中,可以使用修饰核苷酸直接或间接标记 RNA。或者,对扩增的 RNA 进行反转录并标记以生成 cDNA 靶标。
点击图片放大
图9.通过将 RNA 转化为 cDNA,然后从添加的启动子序列进行体外转录,实现 RNA 的扩增。
用于 RNA 测序 (RNA-Seq) 的反转录应用
RNA 测序,又称为 RNA-Seq, 通常可用于研究从基因组转录的 RNA 及其调控机制。随着二代测序 (NGS) 的出现, RNA-Seq 已成为分析全转录组 (即已转录的编码和长链非编码 RNA ) 、检测基因表达、发现剪接变异和融合转录本以及检测低丰度基因的高通量分析方法 [14,15] 。与基因芯片相比,RNA-Seq 的优点包括动态范围更大,灵敏度更高以及可在无基因组信息的情况下表征 RNA 序列。
由于大多数测序平台是为 DNA 设计的,因此 RNA-Seq 的模板制备需要进行反转录。最理想的结果是,所得到的 cDNA 能够无偏差代表初始 RNA,包括低丰度转录物。全长 cDNA 合成对于捕获样本中的所有 RNA 序列也非常重要。反转录的错误率也至关重要,这取决于测序文库大小和数据质量。因此,应谨慎认真选择反转录酶。
了解更多信息:反转录酶特性
研究目标和测序技术将决定 RNA-Seq 模板制备的顺序和方法 [16, 17]。尽管如此,典型的测序文库建库流程包括目标 RNA 的富集、RNA 或 cDNA 的片段化、 反转录 、测序接头 (在多重测序中,还需添加索引或条形码) 以及可选的 文库 PCR 扩增 (图 10)。
图10.RNA 测序的传统实验流程。
了解更多信息:RNA 测序
反转录环介导等温扩增 (RT-LAMP)
RT-LAMP 是一种快速、简单、灵敏的 RNA 检测方案,可使用多种方法用来评估检测结果。由于其具有简单的工作流程和快速的反应时间,尤其适用于在实验室或现场环境中对病毒病原体的检测和监测。
LAMP 方法依赖于具有强链置换活性的 DNA 聚合酶、经特殊设计的内外引物以及环状引物。对于 RNA 靶标的扩增,只需将反转录酶和 RNase 抑制剂添加到 LAMP 反应 (RT-LAMP) 中即可进行一步反应。
LAMP 在恒定温度 (60 – 65°C) 下进行,被归类为等温扩增。LAMP 扩增技术需要 4 或 6 个特殊设计的引物,该引物可与两个不同的靶标区域结合 (相隔~300 bp)。LAMP 最初被开发时使用 4 个引物,随后添加两个环状引物使反应时间缩短了一半。目前,使用 LAMP 法可在 15 分钟内扩增靶标 RNA 或 DNA。LAMP 所需的引物包括两个外部 (F3 和 B3) 引物、两个内部引物 (正向内引物 (FIP) 和反向内引物 (BIP) 和环状引物(正向环状引物 (Loop F) 和反向环状引物 (Loop B))。
LAMP 反应分两步进行:非循环步骤和自动循环步骤。第一步是内引物 (FIP) 与靶标 DNA 结合,开始互补链合成。之后紧接着是外引物 (F3) 的链侵入延伸,释放单链 DNA 作为下游引物的模板。转换得到的内部序列在 F 端形成一个茎环结构。在另一端借助 BIP 引物和 B3 引物进行同样的过程,最终获得在 3′ 和 5′ 端带有茎环的哑铃结构该结构包含多个用于重复扩增的起始位点,并通过自动循环加快了 DNA 扩增,从而得到不同长度和菜花样结构的扩增 DNA (图 11)。
图11.等温 DNA 扩增 (简化示意图) —引物 (3 对) :FIP/BIP、F3/BIP 和 Loop F/B。 靶标序列对茎环的形成比较重要;“ c ”代表互补 (例如,F1c 与 F1 互补)。
RT-LAMP 的优点
- 不需要热循环仪 (仅需加热模块)
- 快速周转时间 (15-60 分钟)
- 多种方法解读检测结果
- 简化的工作流程
- 对抑制剂具有高耐受性
- 适合用于现场检测
RT-LAMP 仅需要少量 RNA,耐受反应抑制剂,易于操作,特异性和灵敏度高。等温条件下的扩增无需热循环仪,扩增效率更高,无需等待温度变化。由于这些特性, LAMP 技术自其发现以来其应用经历了指数级增长。RT-LAMP 在实验室中用于快速检测病原体 (细菌、寄生虫和病毒)。由于其简单性,它也是适应现场或床旁检测的重要方法。
立即查看:LAMP 解决方案
逆转录聚合酶链式反应(RT-PCR)
在RT-PCR中,通过逆转录(RT)将RNA转化为cDNA,然后通过聚合酶链式反应(PCR)扩增cDNA(图1)。利用cDNA扩增步骤,有望对初始RNA进行进一步研究,即便RNA样本数量有限或低丰度表达。RT-PCR的常见应用包括表达基因检测、转录物变异检测,以及克隆和测序用cDNA模板制备。
")
图1. 逆转录聚合酶链式反应(RT-PCR)。RT =逆转录,RTase =逆转录酶
由于逆转录可为PCR扩增和下游实验提供cDNA模板,因此,它是实验成功的关键步骤之一。选定的逆转录酶应对所有样本均具有最高效率,即便是难转录RNA样本,如降解、抑制剂残留或具有高度二级结构的RNA样本。
一步法和两步法是两种最常用的RT-PCR方法,每种方法都具有各自的优缺点(图2)。顾名思义,一步法RT-PCR在单个反应管中将第一链cDNA合成(RT)和后续PCR反应结合在一起。该反应设置可简化工作流程、减少结果差异,并将污染的可能性降至最低。一步法RT-PCR简化了大量样本的处理,适用于高通量应用。但是,一步法RT-PCR采用基因特异性引物进行扩增,将分析局限于每个RNA样本中的几个基因。由于反应需兼顾逆转录和扩增条件,因此,一步法RT-PCR在某些情况下可能具有较低的灵敏度和效率。但是,在RT-PCR中使用基因特异性引物,有助于最大化目标cDNA的得率,并最小化扩增背景。

图2. 一步法和两步法RT-PCR的比较。
两步法RT-PCR包含两个独立反应,首先进行第一链cDNA合成(RT),然后在单个反应管中通过PCR扩增第一步骤中所得cDNA。因此,两步法RT-PCR可用于检测单个RNA样本中的多个基因。RT和PCR反应独立进行,可对每个步骤的反应条件进行优化,使逆转录引物选择(oligo(dT)引物、随机六聚体或基因特异性引物)和PCR反应建立(如DNA聚合酶选择和PCR组分)更灵活。与一步法RT-PCR相比,两步法RT-PCR的缺点包括多个步骤延长了工作流程、增加样本处理和操作步骤以及提高污染和结果变异的可能性。
表1. 对比一步法和两步法RT-PCR
| 一步法RT-PCR | 两步法RT-PCR | |
|---|---|---|
| 反应建立 | 在同时支持逆转录和PCR的条件下,将两个反应结合在一起 | 单独优化的逆转录和PCR反应 |
| 引物 | 基因特异性引物 | oligo(dT)、随机六聚体或基因特异性引物 |
| 最佳用途 | 分析一种或两种基因;高通量平台 | 分析多种基因 |
| 优势 | 方便,高通量 | 灵活 |
定量RT-PCR(RT-qPCR)
定量RT-PCR(RT-qPCR)的最常见应用之一是通过对细胞和组织一段时间或事件后(如,药物治疗)实时mRNA水平的定量分析。RT-qPCR比RT-PCR的灵敏度更高,因此,RT-qPCR也常用于检测研究样本中是否存在逆转录病毒(RNA病毒)。与RT-PCR工作流程相似,RT-qPCR首先将RNA转化为cDNA,然后进行PCR扩增。主要区别在于,RT-qPCR在扩增对数期通过荧光法测定扩增cDNA的水平。扩增水平是对RNA中初始靶标进行定量的基础。(了解关于定量PCR的更多信息)
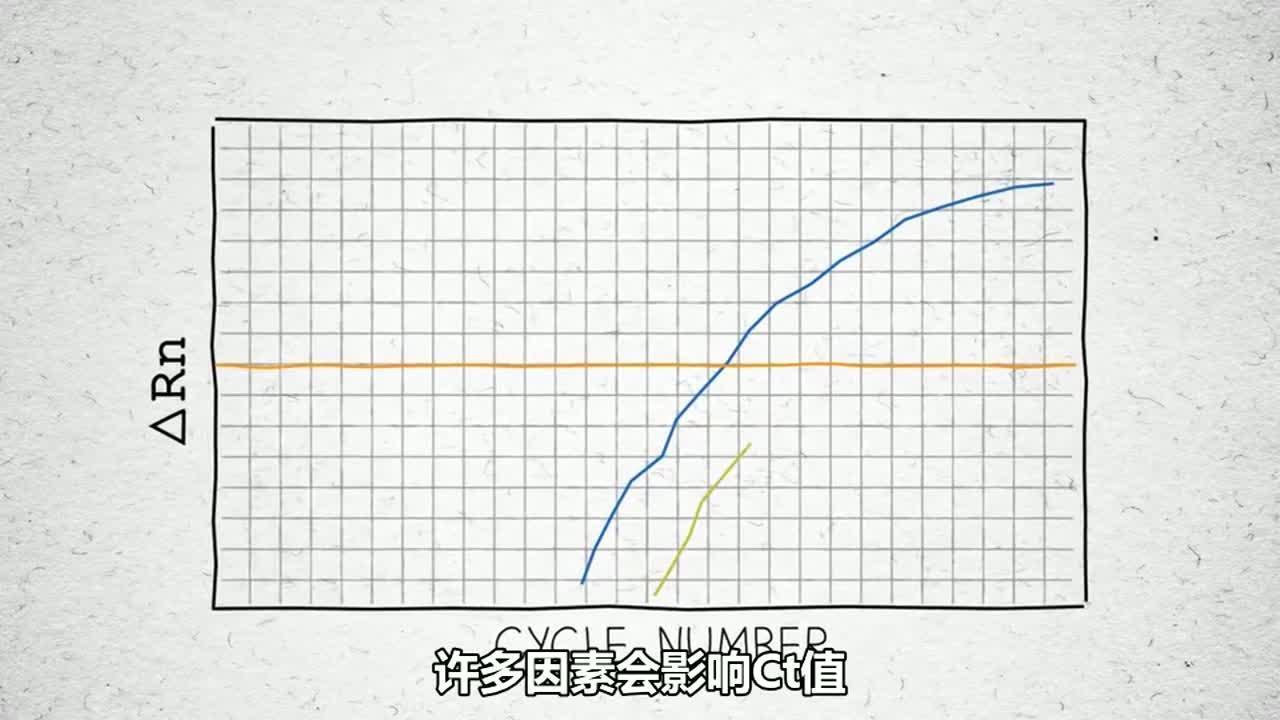
RT-qPCR基因表达定量的准确性在很大程度上取决于cDNA模板的质量和数量。因此,逆转录对于RT-qPCR的成功至关重要。逆转录步骤应产生可代表初始RNA的cDNA产物。因此,所选择的逆转录酶应该具有有效合成cDNA的能力,即使是对低丰度基因以及次优质和/或难转录RNA样本(即富含GC、存在抑制剂或降解RNA样本)。(了解有关逆转录酶属性的更多信息))
除高效逆转录酶之外,在选择逆转录反应试剂方面还需要考虑很多因素。首先,在宽的RNA起始量范围内,cDNA的动态范围或线性扩增是至关重要的。使cDNA得率与RNA起始量成正比,可确保基因表达定量的准确性(图3)。

图3.在总RNA起始量范围内,使用逆转录预混液检测(A)高丰度和(B)低丰度RNA靶标的qPCR线性结果。RNA起始量范围为10 pg至1μg,依次进行逆转录和PCR扩增。两种预混液均产生与RNA起始成比例的cDNA,但是预混液1具有较低(即较早)的Ct值,得率较高,特别是对低丰度基因靶标。
此外,所选试剂在扩增过程中应产生较高且一致的cDNA得率,以获得具有高灵敏度和低变异性的基因表达结果(图4)。单管预混液含有逆转录所需的所有必需组分,有助于将实验变异、交叉污染和移液误差降至最低。(了解有关最佳RT-qPCR逆转录酶的更多信息)

图4.使用不同逆转录预混液检测(A)高丰度和(B)低丰度RNA靶标所得qPCR结果的灵敏度和变异性。在这些试剂中,使用预混液1进行30次重复实验所得结果具有最低的平均Ct值和标准偏差,表明逆转录试剂的选择对于获得可靠的基因表达分析结果非常重要。
RT-qPCR的一个特殊程序是从未经RNA分离的粗细胞裂解物直接进行逆转录[1]。在重点关注稀有细胞或事件的实验中,如使用稀缺样本或选用群体内特定细胞的实验,可考虑使用直接RT-qPCR法来防止可能的样本损失和低RNA回收。在直接RT-qPCR过程中,在细胞裂解过程中抑制可降解RNA的内源性RNA酶和去除细胞基因组DNA是至关重要的步骤。利用优化试剂盒,只需7分钟即可完成样本制备,同时提供来自单个细胞的信号。具有高持续合成能力的逆转录酶具有抑制剂抗性和高灵敏性,特别适用于未纯化RNA提取物的逆转录。
cDNA克隆和文库构建
逆转录酶在分子生物学中的首要应用之一是构建cDNA文库[2-4]。cDNA文库由可代表特定样本中转录序列的cDNA克隆组成。因此,文库提供了关于特定细胞类型、器官或发育阶段的基因时空表达信息。cDNA文库克隆可用于鉴定新型RNA转录物、测定基因序列和重组蛋白的表达。
构建cDNA文库的必要条件是RNA可适当代表其全长和/或相对丰度,因此,逆转录酶的选择非常重要。具有高持续合成能力的逆转录酶可以合成长cDNA,并捕获低丰度RNA。同样,在对具有高度二级结构的RNA进行逆转录时,建议使用热稳定性较强的逆转录酶。(了解关于逆转录特点的更多信息)
在逆转录后,有多种方法可以将cDNA插入克隆载体。第二链合成后获得的双链cDNA通常具有钝端,可以克隆到平末端载体中(图5A)。虽然平末端克隆所含步骤较少,但这种方法可能具有较低的插入效率,并导致插入后的方向性丧失。(了解有关克隆工作流程的更多信息)
或者,可通过修饰在cDNA末端添加具有已知序列的其他核苷酸。例如,为修饰cDNA的5'末端,可使用具有附加5'核苷酸的oligo(dT)引物启动逆转录;为修饰3'末端,可以连接具有目标序列短DNA寡核苷酸(被称为接头或接头)(图5B)。通过这种方式,可将定向插入位点(如限制性和同源重组)、启动子结合(如,T3和T7序列)和亲和纯化(如,生物素和His标签)轻松整合到cDNA序列中。(了解关于DNA文库构建的更多信息)

图5. 常用的cDNA克隆方法。(A)具有平末端的双链cDNA可以直接克隆到平末端克隆载体中。(B)为实现定向克隆,可使用与载体兼容的独特序列修饰cDNA末端。(C)可使用互补末端序列进行无连接克隆,以提高插入效率。(D)若插入序列是已知的,可以考虑通过PCR进行基因特异性克隆。
在另一种常用方法中,使用互补同聚物尾部结构使cDNA插入片段和载体的3'末端酶促延伸。使用末端脱氧核苷酸转移酶(TdT)和单个dNTP,可以在插入片段上添加一串含20-30个核苷酸的序列,并将一串相似的互补核苷酸添加到载体上(如,插入片段上的Cs和载体上的Gs),使载体和插入片段尾部相互退火(图5C)。在转化后,细菌内的间隙被修复,因此不需要连接步骤。
若目标序列是已知的,可通过RT-PCR生成插入片段,用于克隆cDNA的特定区域(图5D)。(了解关于PCR克隆的更多信息)
cDNA末端快速扩增(RACE)
cDNA末端快速扩增(RACE)是一种基于PCR的方法,可用于确定cDNA 5'和3'末端的未知序列[5]。通常,这些方法分别被称为5' RACE和3' RACE。RACE的实验目标包括鉴定5'和3'非翻译区、研究异质性转录起始位点、表征启动子区域、测定完整cDNA序列以及用于蛋白质表达的完整开放阅读框(ORF)的测序。
使用具有单侧特异性的PCR(也称为单侧或锚定PCR [6,7])扩增cDNA的未知区域,用作RACE产物。5' RACE通过延伸具有寡核苷酸的5'末端进行PCR引物结合,而3'RACE利用mRNA的poly(A)尾部结构作为PCR的通用引物位点(图6)。

图6. 5′和3′ RACE。
在5' RACE(图6A)中,使用基因特异性引物,将特定序列或相关家族的mRNA逆转录到第一链cDNA中。然后,使用末端脱氧核苷酸转移酶(TdT)在cDNA 3'末端添加一个同聚物尾部结构(通常是一串Cs),或将cDNA 3'末端连接到寡核苷酸接头上。随后,进行两轮半嵌套PCR,以扩增具有5'未知序列的区域。PCR还可通过下游应用的引物延长扩增子,如定向克隆的限制性位点引入和用于测序的通用测序引物结合位点。
在3′ RACE(图6B)中,使用具有接头序列的oligo(dT)引物将mRNA逆转录成cDNA。然后,使用已知上游外显子序列的特异性引物和通过oligo(dT)引物引入的接头序列,进行两轮半嵌套PCR。通过这种方式,可对外显子和poly(A)尾部结构之间的未知3´ mRNA序列进行扩增,以用于进一步分析。
起始RNA的质量和逆转录反应的设置对于成功完成RACE实验至关重要。在5'RACE中,任何长度的(甚至包括未到达mRNA 5'末端的)第一链cDNA都将具有添加序列(即同聚物尾部结构或接头),随后通过PCR进行扩增。为了最大化全长cDNA的合成,应选择具有最小RNA酶H活性、高持续合成能力和高热稳定性的逆转录酶。(了解有关逆转录酶特点的更多信息)
或者,设计可结合到mRNA 5'末端附近的基因特异性引物,缩短cDNA合成期间与逆转录之间的距离,将有助于捕获未知的5'末端序列。同样,可以考虑在逆转录中选用具有5'-7-甲基鸟苷(7mG)帽子结构的RNA(代表成熟全长真核mRNA)进行程序修饰(图7)[8,9]。

图7. 修饰5'RACE有助于从具有7mG帽子结构的mRNA中捕获序列。首先,使用碱性磷酸酶(AP)从RACE库中去除无帽子结构的RNA,从而去除游离的5'磷酸基团并防止连接。然后,用烟草酸焦磷酸酶(TAP)处理剩余的加帽RNA,去除帽子结构并暴露5'单磷酸盐。然后,将RNA接头连接到暴露的5'磷酸基团,用作5'RACE正向PCR引物的结合位点。
在3'RACE中,全长cDNA并不重要,因为不会扩增PCR起始位点的上游序列。但是,最好选用可生成长cDNA的逆转录酶,因为没有到达PCR引物结合位点的cDNA不会出现在RACE分析中。
基因表达芯片
在20世纪90年代,DNA芯片的发展开辟了大规模无偏差或先前假说的基因表达图谱分析。芯片由玻璃或硅晶片上数千个被称为“features”或“spots”的腔室组成。每个腔室的表面上固定有相同拷贝数的单链DNA序列,称为“探针”,每个探针代表一个基因。探针与用于微阵列的荧光标记cDNA靶标杂交,可同时比较两个样本之间的基因表达(图8和9)[10-12]。

图8. 基因表达微阵列芯片
微阵列探针由生物体基因组或cDNA的已知序列生成。例如,可以利用PCR对每个已知基因进行扩增,然后将其产物变性为单链DNA,并将其固定到芯片上作为探针。或者可直接在芯片上合成20-60nt寡核苷酸,作为芯片探针[13]。
图9概述了如何将基因芯片用于不同样本的基因表达分析。首先,从两个实验(也称为“测试”或“处理”)样本和对照(也称为“参考”或“正常”)样本中分离总RNA或mRNA。然后,将纯化的RNA样本转化为cDNA,并用不同的荧光染料标记。接下来,将两个样本的标记cDNA靶标混合,并与一个微阵列芯片上的探针杂交。洗去未结合的靶标,扫描芯片以检测标记的荧光基团。然后,分析两种荧光信号的比例,从而量化受实验条件影响的基因的表达。

图9. 基因表达芯片的cDNA靶标的制备和分析。
cDNA靶标可在逆转录过程中或之后进行标记(图10)。若采用直接标记,则在cDNA合成期间掺入荧光标记的核苷酸。或者若采用间接标记,可使用修饰的核苷酸进行逆转录,然后使用荧光基团标记cDNA。虽然间接方法需要更长的工作流程,但荧光标记往往更高效[14]。

图10.直接和间接cDNA标记。
当RNA起始量较少时(如10-100ng),可以使用T7-oligo(dT)启动子引物将RNA逆转录为双链cDNA。随后,通过体外转录扩增cDNAs(图11)。在体外转录过程中,可以使用修饰的核糖核苷酸直接或间接标记RNA。或者,对扩增的RNA进行逆转录和标记,生成cDNA靶标[15]。

图11. 通过将RNA转化为cDNA并从附加启动子序列进行体外转录,实现RNA扩增。
在选择逆转录酶以制备用于基因芯片实验的cDNA靶标时,获得高产量全长cDNA的能力对于良好覆盖RNA来说至关重要,包括富含GC或具有二级结构的RNA序列。同样重要的是,为确保获得高信噪比,逆转录酶必须能够有效整合修饰的核苷酸,从而能够准确且无偏差地检测输起始RNA。(了解有关逆转录酶属性的更多信息)
RNA测序(RNA-Seq)
RNA测序,又称为RNA-Seq,通常可用于研究从基因组转录的RNA及其调控作用。随着二代测序(NGS)的出现,RNA-Seq已经成为用于分析全转录组(即转录的编码和长非编码RNA)、测定基因表达、发现剪接变异和融合转录物以及检测低丰度基因的的高通量方法[16,17]。与基因芯片相比,RNA-Seq的优点包括动态范围更大、灵敏度更高以及可在无基因组信息的情况下表征RNA序列。
由于大多数测序平台是为DNA设计的,因此,RNA-Seq模板制备需要进行逆转录。最好的结果是所得cDNA能够无偏差代表初始RNA,包括低丰度转录物。全长cDNA合成对于捕获样本中的所有RNA序列也很重要。逆转录的错误率非常关键,其取决于序列文库的大小和数据质量。因此,应认真选择逆转录酶。(了解有关逆转录酶特点的更多信息)
研究目标和测序技术将决定RNA-Seq模板制备的顺序和方法[18,19]。生成测序文库的标准工作流程包括富集目标RNA、RNA或cDNA片段化、逆转录、测序接头的添加(在多重测序中,还需添加索引或条形码),以及可选的文库PCR扩增(图12)。

图12. RNA测序的传统工作流程。
为了富集mRNA样本,通常需要从样本中去除构成总RNA约80%的核糖体RNA(rRNA),以改善转录组的测序数据。Poly(A)尾部结构通常存在于真核mRNA和长非编码RNA中,所以具有共价结合oligo(dT)的磁珠成为有效富集这些mRNA的替代方法。相比之下,rRNA去除是富集原核mRNA的优选方法,因为原核mRNA不具有可用于分离的Poly(A)尾部结构。对于小RNA(<200nt)来说,可以选用分子量筛选或特殊分离方法。
根据实验目标和测序平台,可选择在逆转录前或逆转录后(即在RNA或双链cDNA上)进行片段化处理。为兼容NGS技术,应制备200-500 nt的片段,以确保高质量读取。片段化方法包括机械法(如超声处理,雾化)、化学法(如水解)和酶促法(如RNase III、DNase I)。
为了检测转录物的方向或正义/反义性(称为“链型”),可以在逆转录之前处理RNA片段,例如使用不同接头标记末端。或者,可在第二链cDNA合成中使用dUTP,从而特异性标记第一链cDNA的互补链(图13)[20]。

图13. 链特异性RNA测序。
片段末端的接头序列可作为与测序平台的连接物。在cDNA合成或扩增过程中,可直接添加或通过特殊设计的引物添加这些接头。除接头外,可以通过PCR加入条形码或索引序列,从而同时对多个样本进行测序(多重测序)。当RNA起始量较低时,PCR有助于生成足够量的输入cDNA用于测序。(了解有关PCR测序的更多信息)
对于测序分析,可使用基因组导向方法或从头方法产生逆转录数据,具体取决于可用的参考基因组类型。基因组导向方法将测序结果映射到已知的基因组序列,而从头方法通过片段重叠组装而得出结果,后者需要大量的计算能力[21]。(了解有关鸟枪法测序的更多信息)
如本节所述,逆转录是cDNA应用工作流程的组成部分。选择最适合您的研究目标的逆转录方法和酶,对于获得成功的实验结果至关重要。
参考文献
- Svec D , Andersson D , Pekny M 等人(2013) 直接细胞裂解用于单细胞基因表达谱分析。Front Oncol 3:274.
- Okayama, H, Berg P (1982) High-efficiency cloning of full-length cDNA.Mol Cell Biol, 2, 2:161-170.
- Gubler, U, Hoffman BJ (1983) A simple and very efficient method for generating cDNA libraries.Gene 25(2-3):263–269.
- Harbers M (2008 年) cDNA 克隆的现状。Genomics 91 (3) 232–242
- Frohman MA , Dush MK , Martin GR (1988) 快速生产稀有转录本中的全长 cDNA :使用单个基因特异性寡核苷酸引物扩增。Proc Natl Acad Sci U S A 85(23):8998–9002.
- Ohara O , Dorit RL , Gilbert W (1989) 单侧聚合酶链反应: cDNA 扩增。Proc Natl Acad Sci U S A 86(15):5673–5677.
- LOH EY , Elliott JF , Cwirla S (1989) 聚合酶单面特异性链反应: T 细胞受体 δ 链分析。Science 243(4888):217-220.
- Maruyama K , Sugano S (1994) 寡核苷酸加盖:一种用寡核糖核苷酸替换真核 mRNA 的帽结构的简单方法。Gene 138(1-2):171–174.
- Schaefer BC (1995) 《 cDNA 末端快速扩增的革命:全长 cDNA 末端聚合酶链反应克隆的新策略》。Anal Biochem 227(2):255-273
- Schena M, Shalon D, Davis RW et al.(1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray.Science 270(5235):467-470.
- Cua DJ, Sherlock J, Chen Y et al.(1999) 使用 cDNA 微阵列进行表达谱分析。Nat Genet 21(1):10–14.
- Capaldi AP (2010) 使用 DNA 微阵列对基因功能进行分析。Methods Enzymol 470:3–17.
- McGall GH ,基督教徒 FC (2002) 高密度 GeneChip 寡核苷酸探针阵列。Adv Biochem Eng Biotechnil 77 : 21 – 42。
- Moravi a , Williams BA , McCue K 等人(2008) 通过 RNA-Seq 对哺乳动物转录组进行映射和定量。Nat Methods 5(7):621–628.
- Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics.Nature Rev Genetics 10 (1) : 57 – 63。
- Van Dijk El , Jaszczyszyn Y , Thermes C (2014) Library Preparation methods for nexion sequencing : Tone down the bias (警告:下一代测序的文库制备方法)。Exp Cell Res, 322, 1:12-20.
- Hrdlickova R , Toloue M , Tian B (2016) RNA-Seq 方法可用于转录组分析。Wiley Interdiscip Rev RNA。 DOI :10.1002/wrna.1364。
Learn more
Explore
- Svec D, Andersson D, Pekny M et al. (2013) Direct cell lysis for single-cell gene expression profiling. Front Oncol 3:274.
- Okayama, H, Berg P (1982) High-efficiency cloning of full-length cDNA. Mol Cell Biol 2(2):161–170.
- Gubler, U, Hoffman BJ (1983) A simple and very efficient method for generating cDNA libraries. Gene 25(2-3):263–269.
- Harbers M (2008) The current status of cDNA cloning. Genomics 91(3):232–242.
- Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci U S A 85(23):8998–9002.
- Ohara O, Dorit RL, Gilbert W (1989) One-sided polymerase chain reaction: the amplification of cDNA. Proc Natl Acad Sci U S A 86(15):5673–5677.
- Loh EY, Elliott JF, Cwirla S (1989) Polymerase chain reaction with single-sided specificity: analysis of T cell receptor delta chain. Science 243(4888):217–220.
- Maruyama K, Sugano S (1994) Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene 138(1-2):171–174.
- Schaefer BC (1995) Revolutions in rapid amplification of cDNA ends: new strategies for polymerase chain reaction cloning of full-length cDNA ends. Anal Biochem 227(2):255–273.
- Schena M, Shalon D, Davis RW et al. (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270(5235):467–470.
- Duggan DJ, Bittner M, Chen Y et al. (1999) Expression profiling using cDNA microarrays. Nat Genet 21(1 Suppl):10–14.
- Capaldi AP (2010) Analysis of gene function using DNA microarrays. Methods Enzymol 470:3–17.
- McGall GH, Christians FC (2002) High-density genechip oligonucleotide probe arrays. Adv Biochem Eng Biotechnol 77:21–42.
- Invitrogen Corp. (2003) Microarray target labeling you can trust. (Brochure)
- Invitrogen Corp. (2004) Comprehensive solutions for microarray analysis. (Brochure)
- Mortazavi A, Williams BA, McCue K et al. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7):621–628.
- Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev Genetics 10(1):57–63.
- van Dijk EL, Jaszczyszyn Y, Thermes C (2014) Library preparation methods for next-generation sequencing: tone down the bias. Exp Cell Res 322(1):12–20.
- Hrdlickova R, Toloue M, Tian B (2016) RNA-Seq methods for transcriptome analysis. Wiley Interdiscip Rev RNA. doi: 10.1002/wrna.1364. [Epub ahead of print]
- Levin JZ, Yassour M, Adiconis X et al. (2010) Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7(9):709–715.
- Kukurba KR, Montgomery SB (2015) RNA Sequencing and Analysis. Cold Spring Harb Protoc 2015(11):951–969.
Share
仅供科研使用,不可用于诊断目的。