Search Thermo Fisher Scientific
逆転写:最も一般的な6つのアプリケーション

RNAは、機能解析のため、逆転写反応(RT)によってより安定な相補DNA(cDNA)へと変換されます。cDNAであれば、クローニング、PCR、シーケンシングといったようなDNAベースの技術が利用できるため、多くのRNAベースの実験ワークフローにおいて、逆転写は極めて重要なステップです。
このページの内容:
逆転写ポリメラーゼ連鎖反応(RT-PCR)
RT-PCRでは、まず逆転写(RT)によってRNAをcDNAに変換し、それからポリメラーゼ連鎖反応(PCR)によってcDNAを増幅します(図1)。RNAの量が限られている、あるいは発現が微量である場合でも、cDNAを増幅することで、元のRNAについて研究を進めることができます。RT-PCRの一般的なアプリケーションとしては、発現遺伝子の検出、転写バリアントの調査、そしてクローニングやシーケンシングのためのcDNAテンプレートの作製などが挙げられます。
")
Figure 1. Reverse transcription polymerase chain reaction (RT-PCR). RT = reverse transcription, RTase = reverse transcriptase.
逆転写反応は、PCR増幅や、それ以降の実験のためのcDNAテンプレートを提供する反応であるため、実験成功の鍵を握る最も重要なステップです。逆転写酵素は、難しいRNAサンプル(例えば、分解している、阻害物質を含んでいる、あるいは高度な2次構造を持っている等)であっても、最も高い反応効率を提供するものでなければなりません。
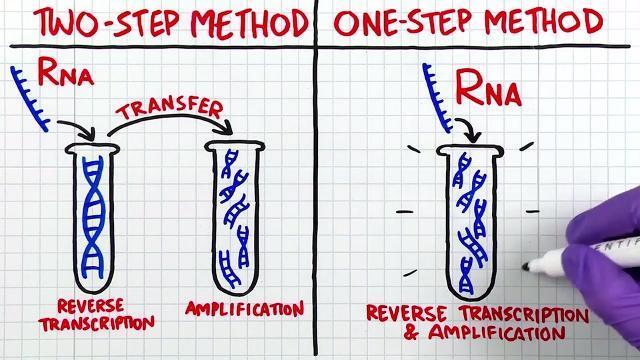
RT-PCRでは、ワンステップ法とツーステップ法の2つの一般的なアプローチがあり、それぞれに良い面と悪い面があります(図2)。ワンステップRT-PCRは、名前の通り、ファーストストランドcDNAの合成(RT)とその次のPCRが1つの反応チューブ内で行われます。この反応セットアップは、ワークフローを簡略化することでばらつきを減らし、またコンタミネーションを最小限にとどめます。ワンステップRT-PCRは、大量サンプルの処理を容易にするため、ハイスループットなアプリケーションに適します。しかし、ワンステップRT-PCRでは増幅の際遺伝子特異的プライマーを使用するため、RNAサンプルあたりの解析遺伝子数は制限されます。ワンステップRT-PCRでは、逆転写反応と増幅反応の条件を妥協し合うことになるため、感度と効率が低くなる傾向にあります。とは言え、遺伝子特異的プライマーの使用は、RT-PCRにおいてターゲットcDNAの収量を最大化し、増幅に伴うバックグラウンドの上昇を最小限にとどめます。

Figure 2. One-step vs. two-step RT-PCR.
ツーステップRT-PCR2つの別々の反応を必要とします。すなわち、ファーストストランドcDNAの合成(RT)をまず行い、その後、生成したcDNAの一部を別のチューブに取り、PCRによって増幅します。そのため、ツーステップRT-PCRは、1つのRNAサンプルで多数の遺伝子を検出したい場合に向いています。RTとPCRを別々に行うことで、それぞれの反応の条件を最適化できるだけでなく、逆転写反応に用いるプライマーの選択(オリゴ(dT)プライマー、ランダムヘキサマー、遺伝子特異的プライマー)と、PCR 設定(DNAポリメラーゼやPCR反応の成分など)の選択をより柔軟に行えます。ワンステップRT-PCRと比べてツーステップRT-PCRの不便な点は、ワークフローに複数の工程や追加のサンプル処理を要することになり、コンタミネーションの危険性や、結果のばらつきが増加してしまう可能性があることです。
表 1.ワンステップRT-PCRとツーステップRT-PCRの比較
| ワンステップRT-PCR | ツーステップRT-PCR | |
|---|---|---|
| セットアップ | 逆転写とPCRの両方に合わせた条件下での反応 | 逆転写とPCRのそれぞれに最適化した反応 |
| プライマー | 遺伝子特異的プライマー | オリゴ(dT)、ランダムヘキサマー、遺伝子特異的プライマーの中から選択 |
| 理想的な使用法 | 1つあるいは2つの遺伝子の解析、ハイスループットプラットフォーム | 複数の遺伝子の解析 |
| 特長 | 簡便、ハイスループット | フレキシブル |
詳細
関連製品
定量RT-PCR (RT-qPCR)
定量RT-PCR (RT-qPCR)の最も一般的なアプリケーションは、mRNAレベルの、経時的変化、あるいは細胞や組織間の違い、または特定のイベント(例えば薬物処理など)における変化、定量的に解析することです。RT-qPCRはRT-PCRよりも感度が高いため、試料中のレトロウイルス(RNAウイルス)の存在を調べる方法としても広く利用されます。RT-PCRのワークフローと同様に、RNAはまずcDNAに変換され、それからPCRによって増幅されます。主な違いは、cDNAレベルを対数増幅期に蛍光によりリアルタイムに測定することです。増幅量が、ターゲットとなるRNAを定量するための基準として使用されます。( 定量 PCRを参照してください)
RT-qPCRによる遺伝子発現定量の正確さは、cDNAテンプレートの質と量に強く依存します。そのため、逆転写のステップが、RT-qPCRの成功の鍵を握ることになります。逆転写では、オリジナルのRNA量比をきちんと反映しているcDNA産物を作製しなければなりません。そのため、使用する逆転写酵素は、たとえ発現量が低く、最適ではなく、難しいRNAサンプルであっても(例えば、GC%が高く、阻害剤が存在し、分解しているなど)、効率良くcDNAを合成できなくてはなりません。(逆転写酵素の特性 を参照してください)
逆転写酵素の反応効率に加えて、RT反応の試薬類の選択についても幾つかの注意点があります。第一に、幅広いインプットRNAに対応できるダイナミックレンジと線形増幅が重要です。インプットRNAの量に比例した収量のcDNAを得ることができる性能は、遺伝子発現の正確な定量を保証します(図3)。

Figure 3. Linearity of qPCR results subsequent to using RT master mixes across a range of total RNA input, for detection of (A) high-abundance and (B) low-abundance RNA targets. RNA input, ranging from 10 pg to 1 μg, was reverse-transcribed and subsequently amplified by PCR. Both master mixes generated cDNA proportional to the input RNA, but a higher yield was obtained from Master Mix 1 as indicated by lower (i.e., earlier) Ct values, especially with the low-abundance gene target.
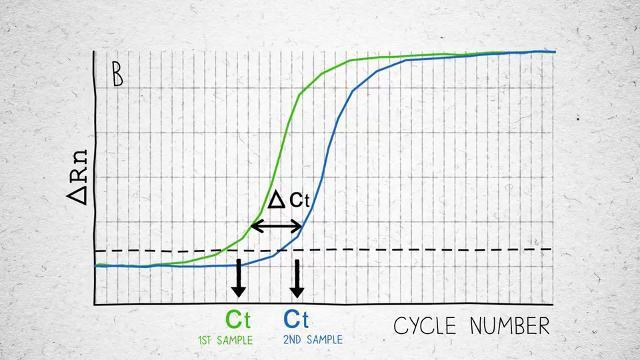
さらに、高感度でばらつきの少ない遺伝子発現解析結果を得るために、選択した試薬類は反復実験において十分量で一貫したcDNA量を生産できなければなりません(図4)。逆転写に必要な成分の全てを含むシングルチューブマスターミックスは、実験間のばらつきや、クロスコンタミネーション、分注エラーなどを最小限に抑えます。(RT-qPCRに最適な逆転写酵素を参照してください)

Figure 4. Sensitivity and variability of qPCR results subsequent to using different RT master mixes, to detect (A) high-abundance and (B) low-abundance RNA targets. Among the reagents, Master Mix 1 produces qPCR results with the lowest average Ct and standard deviation from 30 experimental replicates, demonstrating the importance of reverse transcription reagent choice for reliable gene expression analysis.
RT-qPCRの1つの特殊な方法として、RNAを分離精製せずに、粗細胞溶解液を直接逆転写する方法があります[1]。珍しい細胞やイベントにフォーカスしたり、特定の細胞集団だけを選んだりと、少ないサンプルを用いた実験を行う場合、サンプルロスやRNAの回収量が微量となることを避けるために、ダイレクトRT-qPCRが有効です。ダイレクト法では、RNAを分解し得る内因性のRNaseを阻害することと、細胞内のゲノムDNAを取り除くことが重要です。とともにキットを用いれば、試料の調製が7分ほどで完了する上に、1個の細胞からでもシグナルを得ることができます。未精製RNAの逆転写反応には、高感度で阻害剤に強い高性能の逆転写酵素が特に有効です。

詳細
関連製品
cDNAクローニングとライブラリの構築
分子生物学における逆転写酵素の最初のアプリケーションの1つは、cDNAライブラリの構築でした[2-4]。cDNAライブラリは、特定の試料に含まれる転写産物を反映したcDNAクローンで構成されます。そのため、ライブラリは、細胞の種類、臓器、発達段階などに依存した時間的かつ空間的な遺伝子発現情報を提供します。cDNAライブラリのクローンは、新規RNAの解析や遺伝子配列の決定、あるいは組み換えたんぱく質の発現などに利用されます。
cDNAライブラリの構築に不可欠な要素は、RNAの鎖長や相対量をきちんと反映していることであり、そのためには、逆転写酵素の選択が極めて重要です。高性能の逆転写酵素は、存在量の少ないRNAだけでなく、鎖長の長いcDNAを合成することもできます。同様に、耐熱性を向上させた逆転写酵素は、高度な2次構造を持ったRNAを逆転写するのに推奨されます。(逆転写酵素の特性 を参照してください)
逆転写の後、クローニングのためにベクターにcDNAを挿入する方法が幾つかあります。セカンドストランド合成を終えた2本鎖cDNAは、しばしば平滑末端を持つことがあるので、平滑末端ベクターでクローニングできます(図5A)。この平滑末端クローニングは、工程は少ないですが、挿入後のライゲーション効率が低く、方向性が損なわれることがあります。(クローニングワークフロー を参照してください)
あるいは、cDNA末端を既知配列のヌクレオチドで修飾する方法もあります。例えば、cDNAの5′末端を修飾する場合は、5′末端に追加のヌクレオチドを持ったオリゴ(dT)プライマーを逆転写の際に使用し、3′末端を修飾する場合は、任意の配列を持ったリンカーやアダプターと呼ばれる短いDNAオリゴをライゲーションします(図5B)。このやり方法で、方向性を持たせた挿入や(制限酵素を用いた方法、もしくは相同組み換えなど)、プロモーターの結合(T3やT7配列など)、そしてアフィニティ精製(ビオチンやHisタグなど)のためのサイトを簡単にcDNA配列の中に組み込むことができます。(DNAライブラリの構築を参照してください)

Figure 5. Common methods to clone cDNA. (A) Double-stranded cDNA with blunt ends may be cloned directly into a blunt-end cloning vector. (B) For directional cloning, cDNA ends can be modified with unique sequences compatible with the vector. (C) Ligation-independent cloning may be performed with complementary terminal sequences to enhance the efficiency of insertion. (D) Gene-specific cloning via PCR may be considered when the insert’s sequence is known.
別の一般的な方法としては、挿入するcDNAとベクターの3′末端を、相補的なホモポリマーで酵素的に伸長するやり方が挙げられます。ターミナルデオキシヌクレオチド転移酵素(TdT)と1種類のdNTPを用いて、20~30ほどの連なったヌクレオチドを遺伝子に付加し、それと相補的なヌクレオチドを同様にベクターに付加することで(例えば、挿入遺伝子に連続したCを、ベクターに連続したGを)、ベクターと挿入遺伝子のそれぞれのテールをアニーリングさせることができます(図5C)。形質転換の後、細菌の中でギャップは修復されるため、ライゲーションする必要はありません。
cDNAの特定の領域のクローニングにおいて、ターゲットとなる遺伝子配列が分かっている場合は、挿入遺伝子をRT-PCRによって作製できます(図5D)。(PCRクローニング を参照してください)
詳細
関連製品
RACE(Rapid amplification of cDNA ends)
RACE(Rapid amplification of cDNA ends)は、cDNAの5′末端と3′末端の未知配列の決定に特化した、PCRベースの方法です[5]。この方法は一般的に、5′ RACEおよび3′ RACEとして知られています。RACEの目的としては、5′末端と3′末端の非翻訳領域の同定、ヘテロな転写開始部位の研究、プロモーター領域の解析、cDNAの完全配列の決定、そしてタンパク質発現のための完全なオープンリーディングフレーム(ORF)のシーケンシング、などが挙げられます。
RACEの生成物としてcDNAの未知領域を増幅するために、片側に特異的なPCRが用いられます(ワンサイド、あるいはアンカードPCRとも呼ばれます[6,7])。5′ RACEはPCRプライマーがアニーリング可能なオリゴヌクレオチドを5′末端に付加する必要がありますが、3′ RACEはPCRでの一般的なプライミングサイトであるmRNAのポリ(A)テールが使用できます(図6)。

Figure 6. 5′ and 3′ RACE.
5′ RACE(図6A)では、特定の配列やそれに関連するファミリーのmRNAが、遺伝子特異的プライマーによってファーストストランドcDNAに逆転写されます。その後cDNAの3′末端に、ターミナルデオキシヌクレオチドトランスフェラーゼ(TdT)によってホモポリマーテール(通常は連続したC)付加するか、あるいはオリゴヌクレオチドアダプターがライゲーションされます。さらに、5′末端の未知配列領域を増幅するために、2ラウンドのネステッドPCRが行われます。このPCRにより、方向性クローニングに用いる制限酵素切断配列等や、シーケンシングのためのユニバーサルプライマーアニーリングサイトのような、ダウンストリームアプリケーションのための配列の付加が可能です。
3′ RACE(図6B)では、アダプターシーケンスを持ったオリゴ(dT)プライマーによって、mRNAがcDNAに逆転写されます。それから、既知のエクソン配列とオリゴ(dT)プライマーによって導入されたアダプター配列に特異的なプライマーによって、2ラウンドのネステッドPCRが行われます。このようにして、エクソンとポリ(A)テールの間の未知の3´ mRNA配列が増幅され、解析に用いられます。
インプットRNAの質と逆転写反応のセットアップが、RACE実験における成功の鍵を握っています。5′ RACEでは、いかなる長さのファーストストランドcDNAも(すなわちmRNAの5′末端に達していなくても)付加配列(つまりホモポリマーテールやアダプター)を持っており、その後、PCRで増幅されます。cDNA合成をできるだけ完全長にするためには、RNase H活性を最小化した、高性能で高耐熱性の逆転写酵素を選択する。(逆転写酵素の特性 を参照してください)
また、mRNAの5′末端に近い位置に結合する遺伝子特異的プライマーの設計は、cDNAの合成段階で逆転写の距離が短くなるため、未知の5′末端配列取得を容易にします。同様に、成熟した真核生物の完全長mRNAに存在する5′ 7-メチルグアノシン(7mG)キャップを利用しても良いかもしれません(図7)[8,9]。

Figure 7. Modified 5′ RACE to help capture sequences from 7mG-capped mRNA. First, uncapped RNA is eliminated from the RACE pool by using alkaline phosphatase (AP), to remove the free 5′ phosphate group and prevent ligation. Then, the remaining capped RNA is treated with tobacco acid pyrophosphatase (TAP) to remove the cap and expose the 5′ monophosphate. An RNA adapter is then ligated to the exposed 5′ phosphate group and serves as a binding site for the forward PCR primers of 5′ RACE.
3′ RACEでは、PCRプライミングサイトの上流に位置する配列は増幅されないため、cDNAは完全長である必要はありません。とは言え、PCRプライマー結合部位に届かないcDNAではRACE分析に利用できないため、長鎖のcDNAを生成できる逆転写酵素が好ましいです。
詳細
関連製品
遺伝子発現マイクロアレイ
1990年代のDNAマイクロアレイの開発は、バイアスや仮説を排除した、遺伝子発現大規模プロファイリングを可能にしました。マイクロアレイは、ガラスあるいはシリコンウエハ上の「フィーチャー」や「スポット」として知られる、数千のチャンバーからなります。各フィーチャーは、各遺伝子用に設計された「プローブ」と呼ばれる1本鎖DNAの同一コピーが表面に固定化されたものです。プローブはマイクロアレイにアプライされた蛍光標識cDNAターゲットとハイブリッドを形成します。この方法では、サンプル間の遺伝子発現を、同一チップ上で同時に比較することができます(図8と図9)[10-12]。

Figure 8. Gene expression microarray chip.
マイクロアレイプローブは、対象生物のゲノムあるいはcDNAの既知の配列から作製します。例えば、全ての既知遺伝子のコピーをPCRにより作製し、生成物を1本鎖DNAに変性してから、固定化プローブとしてチップ上にスポットします。または、プローブとなる20~60個のヌクレオチドをチップ上で直接合成することも可能です[13]。
図9は、マイクロアレイの遺伝子発現ディファレンシャル解析の概要を示しています。最初に、トータルRNAやmRNAを2つのサンプル、つまり検体(「テスト」や「処理後」とも呼ばれます)と、コントロール(「リファレンス」や「ノーマル」とも呼ばれます)から単離します。精製RNAサンプルを、cDNAに変換し、それぞれ異なる蛍光色素で標識します。次に、両サンプルの標識cDNAを混合し、一枚のマイクロアレイチップ上のプローブにハイブリダイズします。結合しなかったターゲットを洗浄後、標識蛍光を検出するため、マイクロアレイをスキャンします。その後、実験条件による遺伝子発現の変化を定量化するため、2つの蛍光シグナルの比率を解析します。

Figure 9. Preparation and analysis of cDNA targets for gene expression microarrays.
cDNAターゲットの標識は、逆転写の最中、あるいは後のどちらでも可能です(図10)。直接標識法では、蛍光標識ヌクレオチドをcDNA合成の際に取り込ませます。あるいは、後で標識しやすいように修飾したヌクレオチドを逆転写に利用し、その後、得られたcDNAを蛍光色素で標識することも可能です。間接法では長いワークフローを必要としますが、蛍光標識効率は一般的に優れています[14]。

Figure 10. Direct and indirect cDNA labeling.
インプットRNAの量が少ない場合は(例えば10~100 ng)、まずT7-オリゴ(dT)プロモータープライマーを利用して、RNAから2本鎖cDNAを合成します。そうして得られたcDNAをin vitro転写によって増幅します(図11)。in vitro転写の際、RNAは修飾リボヌクレオチドによって直接、あるいは間接的にラベル化できます。または、増幅したRNAを逆転写する際に標識化することも可能です[15]。

図 11.cDNA合成、in vitro転写によるRNAの増幅
RNAのポピュレーションを正確に反映させるためには、マイクロアレイ実験に使用するcDNAターゲットの調製のための逆転写酵素は、高GC含量、あるいは2次構造を形成するRNAであっても完全長cDNAを高収率で得ることのできる能力を持っていることが重要です。同じく重要な点として、正確でバイアスのないインプットRNAの検出を可能にする高いシグナル/バックグラウンド比率を得るため、逆転写酵素は修飾ヌクレオチドを効率良く取り込めなければなりません。(逆転写酵素の特性 を参照してください)
詳細
関連製品
RNAシーケンシング(RNA-Seq)
RNAシーケンシング(RNA-Seq)は、一般的に、ゲノムから転写されるRNAとその転写調節に関する情報を得るために行われます。次世代シーケンシング(NGS)の出現とともにRNA-Seqは、全体のトランスクリプトーム(すなわち、翻訳されたコーディング、および長鎖ノンコーディングRNA)の解析、発現遺伝子の決定、スプライスバリアントや融合転写産物の発見、そして存在比の少ない遺伝子の検出、などのためのハイスループットな方法として確立されてきました[16,17]。RNA-Seqはマイクロアレイと比べると、非常にダイナミックレンジが広く、高感度で、予備的なゲノム情報なしにRNA配列を解析することができるといった利点を持ちます。
大抵のシーケンシングプラットフォームはDNA用にデザインされているため、RNA-Seqのためのテンプレートの調製は逆転写反応を伴います。作製されたcDNAは、存在量の少ないものも含まれますが、できる限りバイアスのない、オリジナルRNAを反映していることが望まれます。サンプル中の全てのRNA配列を対象とするためには、合成cDNAが完全長かどうかも重要になります。シーケンシングライブラリのサイズやデータの品質によっては、逆転写のエラー率が重要になってきます。そのため、使用する逆転写酵素を慎重に選択しなければなりません。(逆転写酵素の特性 を参照してください)
新たな研究目的、シーケンシング技術の進歩は、そのためのテンプレート調製の方法を要求します[18,19]。しかしながら、シーケンシングのためのライブラリ作製における典型的なワークフローは、目的RNAの濃縮、RNAやcDNAの断片化、逆転写、シーケンシングアダプターの付加(マルチプレックスであれば、インデックスやバーコードも)、そしてオプションとしてのライブラリのPCR増幅、といった多くのプロセスを含みます(図12)。

Figure 12. Traditional workflow of RNA sequencing.
mRNAの濃縮では、トランスクリプトームのシーケンシングデータをより良いものにするため、トータルRNAの約80%であるリボソームRNA(rRNA)をサンプルから除去します。真核生物のmRNAと長鎖ノンコーディングRNAにはポリ(A)テールが存在するため、オリゴ(dT)を共有結合させた磁気ビーズが、そのようなmRNAの効率的な濃縮に使用できます。一方、原核生物のmRNAは分離に利用できるポリ(A)テールを持たないため、それらの濃縮にはrRNAを除去する方法が選択されます。スモールRNA(<200 nt以下)には、サイズ選択法または特別な分離法が、代わりに用いられます。
実験の目的とシーケンシングのプラットフォームによっては、逆転写反応の前、あるいは後に(すなわちRNAや2本鎖cDNAに対して)断片化が行われます(図12と13<)。高品質な解読を確実にするため、NGS技術に適した200~500 ntの断片を調製します。断片化の手段としては、機械的(例えば、ソニケーションやせん断など)、化学的(加水分解など)、そして酵素的(例えばRNase IIIやDNase Iなど)な方法などが挙げられます。
転写産物の方向性やセンス/アンチセンスの特徴(「ストランデッドネス」と呼ばれる)を調べるためには、区別化アダプターによる末端修飾のように、逆転写に先立ちRNA断片を処理しておきます。また、ファーストストランドcDNAの相補鎖に特異的に印を付けられるように、セカンドストランドcDNAの合成の際にdUTPを使用する方法もあります(図13)[20]。

Figure 13. Strand-specific RNA sequencing.
断片の末端のアダプター配列は、シーケンシングに役立ちます。こういったアダプターは、cDNA合成や増幅の間に、直接、あるいは、特別にデザインされたプライマーによって付加されます。アダプターに加え、バーコードやインデックス配列を、同時かつ複数サンプルのシーケンシング(マルチプレキシング)のために、PCRの際に取り込ませることもできます。使用できるRNAの出発量が少ない時には、シーケンシングに適した量のインプットcDNAをPCRによって調製します。(シーケンシングにおけるPCR を参照してください)
シーケンシング解析では、ゲノム情報が利用可能かどうかにもよりますが、ゲノム情報を基にした、あるいはde novo法による、トランスクリプトームデータが利用できます。ゲノム情報を基にした方法では、得られた配列結果を既知のゲノム配列にマッピングします。一方、de novo法では、大規模なコンピューターによるコンティグのアセンブリから結果を引き出します[21]。(ショットガンシーケンシング を参照してください)
本項で述べたように、逆転写は、cDNAベースのアプリケーションにおけるワークフローに不可欠です。研究目的に最も適している逆転写の方法や酵素の選択が重要です。
詳細
関連製品
参考文献
- Svec D, Andersson D, Pekny M et al.(2013) Direct cell lysis for single-cell gene expression profiling.Front Oncol3:274.
- Okayama, H, Berg P (1982) High-efficiency cloning of full-length cDNA.Mol Cell Biol2(2):161–170.
- Gubler, U, Hoffman BJ (1983) A simple and very efficient method for generating cDNA libraries.Gene25(2-3):263–269.
- Harbers M (2008) The current status of cDNA cloning.Genomics91(3):232-242.
- Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer.Proc Natl Acad Sci U S A85(23):8998-9002.
- Ohara O, Dorit RL, Gilbert W (1989) One-sided polymerase chain reaction: the amplification of cDNA.Proc Natl Acad Sci U S A86(15):5673-5677.
- Loh EY, Elliott JF, Cwirla S (1989) Polymerase chain reaction with single-sided specificity: analysis of T cell receptor delta chain.Science243(4888):217-220.
- Maruyama K, Sugano S (1994) Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides.Gene138(1-2):171-174.
- Schaefer BC (1995) Revolutions in rapid amplification of cDNA ends: new strategies for polymerase chain reaction cloning of full-length cDNA ends.Anal Biochem227(2):255-273.
- Schena M, Shalon D, Davis RW et al.(1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray.Science270(5235):467-470.
- Duggan DJ, Bittner M, Chen Y et al.(1999) Expression profiling using cDNA microarrays.Nat Genet21(1 Suppl):10–14.
- Capaldi AP (2010) Analysis of gene function using DNA microarrays.Methods Enzymol470:3-17.
- McGall GH, Christians FC (2002) High-density genechip oligonucleotide probe arrays.Adv Biochem Eng Biotechnol77:21–42.
- Invitrogen Corp. (2003) Microarray target labeling you can trust.(Brochure)
- Invitrogen Corp. (2004) Comprehensive solutions for microarray analysis.(Brochure)
- Mortazavi A, Williams BA, McCue K et al.(2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq.Nat Methods5(7):621-628.
- Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics.Nature Rev Genetics10(1):57-63.
- van Dijk EL, Jaszczyszyn Y, Thermes C (2014) Library preparation methods for next-generation sequencing: tone down the bias.Exp Cell Res322(1):12-20.
- Hrdlickova R, Toloue M, Tian B (2016) RNA-Seq methods for transcriptome analysis.Wiley Interdiscip Rev RNA. doi: 10.1002/wrna.1364.[Epub ahead of print]
- Levin JZ, Yassour M, Adiconis X et al.(2010) Comprehensive comparative analysis of strand-specific RNA sequencing methods.Nat Methods7(9):709-715.
- Kukurba KR, Montgomery SB (2015) RNA Sequencing and Analysis.Cold Spring Harb Protoc2015(11):951-969.
Share
For Research Use Only. Not for use in diagnostic procedures.